The Birth, Life, and Death of Red Blood Cells: Erythropoiesis, The Mature Red Blood Cell, and Cell Destruction
John G. Quigley
Robert T. Means, Jr.
Bertil Glader
ERYTHROPOIESIS
Concept of the Erythron
“There is, unfortunately, no name for this tissue (or organ), and it will save a good deal of paraphrasing and probably some confusion if we make one and call it erythron.”1
The entire process by which red cells are produced in the bone marrow is termed erythropoiesis. For descriptive purposes, the process can be divided into various stages, including the commitment of pluripotent stem cell progeny to erythroid differentiation, the erythropoietin (Epo)-independent or early phase of erythropoiesis, and the Epo-dependent or late phase of erythropoiesis. Under normal conditions, erythropoiesis results in a red cell production rate such that the red cell mass in the body remains constant, indicating the presence of regulatory control mechanisms. The control mechanisms regulating the later phases of erythropoiesis are better understood than those regulating the early phases. The hormone Epo is established as the major factor governing red cell production.2,3 and 4 Erythropoiesis involves a great variety and number of cells at different stages of maturation, from stem cell progeny committed to erythroid differentiation to the mature circulating red cell (Fig. 6.1). The whole mass of these erythroid cells has been termed the erythron,1 a concept that emphasizes the functional unity of the red cells, their morphologically recognizable marrow precursors, and the functionally defined progenitors of these precursors. The concept of erythron as a tissue has thus far contributed significantly to the understanding of the physiology and pathology of erythropoiesis.
ERYTHROID CELLS
Committed Erythroid Progenitors
The processes leading from the undifferentiated hematopoietic stem cell to erythroid commitment are discussed in Chapter 5. Erythroblasts in the bone marrow are generated from proliferating and differentiating earlier, more immature erythroid cells termed erythroid progenitors. These progenitor cells are detectable functionally by their ability to form in vitro erythroid colonies.5 The development of tissue culture techniques for cloning hematopoietic progenitor cells in semisolid culture media in vitro has led to the recognition and assay in the human and murine bone marrow of at least two erythroid progenitors, the colony-forming unit-erythroid (CFU-E) and the burst-forming unit-erythroid (BFU-E). Under the influence of Epo, these progenitors can grow in semisolid culture media and give rise to colonies of well-hemoglobinized erythroblasts. A two-phase liquid culture system that models human erythroid development has also been described.6
Colony-forming Unit-Erythroid
The CFU-E is an erythroid cell closely related to the proerythroblast.7 Under the influence of low concentrations of Epo, it gives rise (in 2 to 3 days in murine and in 5 to 8 days in human marrow) to colonies of 8 to 32 well-hemoglobinized cells7,8,9 and 10 (Fig. 6.2). The clonal origin of these colonies has been demonstrated by glucose-6-phosphate dehydrogenase-isoenzyme analysis.11 Morphologically, CFU-E purified from progenitor cell cultures appear as immature cells with fine nuclear chromatin; a well-defined, large nucleolus; a high nuclear-cytoplasmic ratio; a perinuclear clear zone; and basophilic cytoplasm with pseudopods.12,13 On electron microscopy, this cell appears as a primitive blast with dispersed nuclear chromatin, a prominent nucleolus, and an agranular cytoplasm containing clumps of mitochondria and frequent pinocytotic vesicles.12 The number of CFU-E in the human marrow ranges from 50 to 400/105 light-density, nonadherent, nucleated cells and varies significantly with the methods used for cell separation and the culture conditions. The majority of CFU-E are in a phase of active DNA synthesis (S phase) as demonstrated by a 70% to 90% killing of cells after short exposure to 3H-thymidine in vitro (3H-thymidine suicide) or after administration of cycle-specific chemotherapeutic agents in vivo.14,15 and 16 The size of the CFU-E compartment in intact animals depends on the levels of circulating Epo. Anemia associated with high Epo levels or the administration of Epo leads to expansion of the CFU-E compartment, whereas transfusion-induced polycythemia leads to low Epo levels and a significant reduction of the CFU-E compartment.16 From a number of in vitro studies, it has been established that the CFU-E is the most Epo-sensitive cell, carrying the highest density of Epo receptors (EpoR) on its surface, and it is also absolutely dependent on Epo for its survival. In the absence of Epo, CFU-E rapidly undergoes programmed cell death (apoptosis)12,17,18,19,20 Although the first phase of CFU-E differentiation is Epo-dependent, the later stages are not.21
Highly purified CFU-E has been isolated from murine fetal liver cells; this is defined in flow cytometry studies as expressing c-kit, the receptor for the cytokine stem cell factor (SCF), negative for a number of lineage markers (Ter119, B220, Mac-1, CD3, Gr-1, CD32/16, Sca-1, and CD41), and having high cell surface expression of the transferrin receptor (TfR) (CD71) and the heat stable antigen, CD24.13
Burst-forming Unit-Erythroid
The BFU-E is an erythroid progenitor that is much more immature than the CFU-E, and more closely related to the multipotent hematopoietic stem cell, as indicated by its cell size, buoyant density, and the relatively low percentage of these cells in active DNA synthesis (0% to 25%).15,16,22,23 In contrast to CFU-E, BFU-E also has a (limited) capacity for self-renewal and is detectable in
the peripheral blood at a concentration of 0.02% to 0.05% of lightdensity mononuclear blood cells.24,25 BFU-E can be separated from CFU-E by its slower velocity sedimentation at unit gravity.26 Morphologically, the BFU-E appears as a very immature blast cell with slightly oval, moderately basophilic cytoplasm with occasional pseudopods, very fine nuclear chromatin, and large nucleoli.13,19 On electron microscopy, the cytoplasm contains polyribosomes and is less abundant than in CFU-E, whereas the nucleus contains small amounts of clumped heterochromatin and prominent nucleoli.19
the peripheral blood at a concentration of 0.02% to 0.05% of lightdensity mononuclear blood cells.24,25 BFU-E can be separated from CFU-E by its slower velocity sedimentation at unit gravity.26 Morphologically, the BFU-E appears as a very immature blast cell with slightly oval, moderately basophilic cytoplasm with occasional pseudopods, very fine nuclear chromatin, and large nucleoli.13,19 On electron microscopy, the cytoplasm contains polyribosomes and is less abundant than in CFU-E, whereas the nucleus contains small amounts of clumped heterochromatin and prominent nucleoli.19
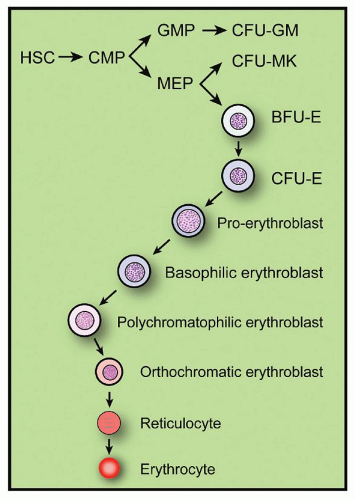 FIGURE 6.1. Schematic representation of the differentiation of erythroid cells from multipotent hematopoietic stem cells. BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid; CFU-GM, colony-forming unit-granulocyte-monocyte; CFU-MK, colony-forming unit-megakaryocyte; CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; MEP, megakaryocyte/erythroid progenitor. |
In the presence of Epo and under the influence of other cytokines that act on early hematopoietic cells, such as interleukin-3 (IL-3), IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), thrombopoietin (Tpo), and SCF, the BFU-E progenitor gives rise in 5 to 7 days in mice and in 14 to 16 days in humans to clusters of many erythroid colonies (a large “burst”) containing a total of 500 to more than 30,000 to 40,000 well-hemoglobinized erythroblasts (Fig. 6.3). Cytokines such as transforming growth factor (TGF) β, tumor necrosis factor (TNF) α, and interferon (IFN) γ, on the other hand, suppress progenitor proliferation. The clonal origin of BFU-E-derived erythroblasts has been demonstrated by characterization of the type of hemoglobin produced by cells in single colonies in co-culture experiments of bone marrow cells from a patient with homozygous hemoglobin C and marrow cells from another patient with homozygous hemoglobin S disease.27 The BFU-E can be considered as a progenitor of the CFU-E. Indeed, after 6 to 8 days in culture, cells generated from human BFU-E have all the functional characteristics of CFU-E.12 The concentration of BFU-E in human bone marrow varies from 10 to 50/105 nucleated cells; however, this number fluctuates widely depending on the cell separation methods and culture conditions. From both in vitro and in vivo experiments, it has been established that the early stages of BFU-E proliferation and differentiation are Epo independent.16,19,23 BFU-E can survive in vitro for 48 to 72 hours in the absence of Epo, but it is absolutely dependent on IL-3 for survival.19 Only 20% of blood BFU-E expresses a very low density of EpoR detectable by autoradiography.19 The size of the BFU-E compartment in the marrow of animals remains unaffected by the acute changes in the levels of circulating Epo induced by anemia or transfusional polycythemia.16 Anemia can induce an increase in the cycling of BFU-E without affecting their numbers,28 and in vitro Epo can induce BFU-E into DNA synthesis.29 In humans, chronic administration of Epo is associated with an increase in the concentration and cycling status of marrow BFU-E; however, these changes are also seen in granulocytic -monocytic and megakaryocytic CFUs (CFU-GM, CFU-MK), and multilineage progenitors such as CFU-GEMM, indicating that, at the early progenitor cell level, the marrow responds to Epo as an organ in a non-lineage-specific manner.30 All these evidences indicate that the early stages of erythropoiesis at the BFU-E level are Epo independent, and Epo dependence develops at a stage between BFU-E and CFU-E.19 The distinction between early (BFU-E) and late (CFU-E) erythroid progenitors, although valid, is by itself artificial. There is a variety of cells
between BFU-E and CFU-E that form a continuum of erythroid progenitors at different stages of differentiation with properties between those of BFU-E and CFU-E. As an example, a subclass of erythroid progenitors termed mature BFU-E has been described in human and murine marrow.14,15 These cells share properties of both CFU-E and BFU-E. They have a proliferative potential lower than BFU-E but higher than CFU-E, their cycling status is also intermediate between CFU-E and BFU-E, and they do not exhibit IL-3 dependence, but show relative Epo dependence.14,15,18 Thus, the evidence suggests that during erythroid development, early progenitors of high proliferative potential in a relatively low cycling status with absolute dependence on IL-3 and responsiveness to, but not dependence on, Epo differentiate progressively through various stages into later progenitors of low proliferative potential with a high cycling status that are IL-3 independent and totally Epo dependent.
between BFU-E and CFU-E that form a continuum of erythroid progenitors at different stages of differentiation with properties between those of BFU-E and CFU-E. As an example, a subclass of erythroid progenitors termed mature BFU-E has been described in human and murine marrow.14,15 These cells share properties of both CFU-E and BFU-E. They have a proliferative potential lower than BFU-E but higher than CFU-E, their cycling status is also intermediate between CFU-E and BFU-E, and they do not exhibit IL-3 dependence, but show relative Epo dependence.14,15,18 Thus, the evidence suggests that during erythroid development, early progenitors of high proliferative potential in a relatively low cycling status with absolute dependence on IL-3 and responsiveness to, but not dependence on, Epo differentiate progressively through various stages into later progenitors of low proliferative potential with a high cycling status that are IL-3 independent and totally Epo dependent.
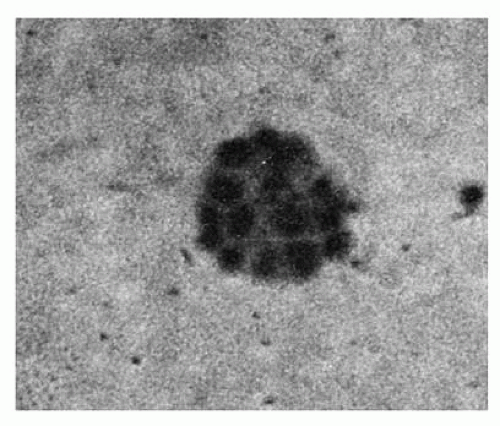 FIGURE 6.2. A day 7 colony-forming unit-erythroid-derived colony of erythroblasts containing 16 cells. |
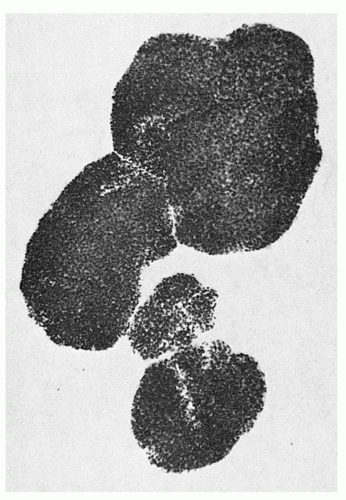 FIGURE 6.3. A day 15 human bone marrow burst-forming unit-erythroid-derived burst (group of colonies) of erythroblasts containing over 1,000 cells. |
Increasingly, in place of functional definition of erythroid progenitors, studies, especially in mice but also in humans, allow for prospective identification of progenitors using cell surface markers.31,32 As noted above, recent work, for example, using murine fetal liver cells as a source of hematopoietic—at the fetal liver stage predominantly erythroid—cells allows isolation of BFU-E and CFU-E progenitors with 94% and 95% purity, respectively: although both these erythroid progenitors express c-kit, BFU-E is distinguished from CFU-E as it has much lower cell surface expression of CD24 and CD71.13
Erythroid Precursors
The least mature recognizable erythrocyte precursor cell is known as the proerythroblast (or pronormoblast). The various stages of maturation, in order of increasing maturity, are proerythroblasts, basophilic erythroblasts, polychromatophilic erythroblasts, and orthochromatic erythroblasts. The morphologic characteristics of each stage, as seen with light microscopy after staining with Romanowsky dyes, are widely agreed upon. Cytoplasmic maturation is assessed by the change in staining characteristics, as the deep blue color derived from the high RNA content of immature cells gives way to the red color characteristic of hemoglobin. Nuclear maturation is evaluated by the disappearance of nucleoli and the condensation of chromatin as nuclear activity decreases. In addition, there is a gradual decrease in cell size and EpoR expression and terminally exit from the cell cycle.
Stages of Erythroblastic Differentiation
The proerythroblast is a round or oval cell of moderate to large size (14 to 19 µm diameter; Figs. 6.4A and 6.5A). It possesses a relatively large nucleus, occupying perhaps 80% of the cell, and a rim of basophilic cytoplasm. The nucleus of the youngest cells in this group differs little from that of the myeloblast. Nucleoli are seen and may be prominent. Only small amounts of hemoglobin are present that cannot be detected by Giemsa stain. As compared with that of myeloblasts and lymphoblasts, the cytoplasm is more homogeneous and condensed and may appear granular. A small pale area may be found in the cytoplasm, probably corresponding to the Golgi apparatus.33 The nuclear chromatin is somewhat coarser than that in myeloblasts or lymphoblasts.
The basophilic erythroblast is similar to the proerythroblast except that nucleoli are no longer visible and the cell is smaller (12 to 17 µm in diameter; Figs. 6.4B and 6.5B). Condensation of chromatin (formation of heterochromatin) begins and, on light microscopy, the chromatin may appear coarse and granular; thus, there is little resemblance to the myeloblast. The nuclear structure may assume a wheel-spoke arrangement. Ribosomes reach their maximum number during this stage, and thus the cytoplasm is deeply basophilic. Cytoplasmic color changes during subsequent stages reflect the increase in acidophilic hemoglobin and the decreasing amount of ribosomal RNA.
The first faint blush of hemoglobin, as indicated by one or more pink areas near the nucleus in dry fixed preparations, introduces the next stage, the polychromatophilic erythroblast (Figs. 6.4C and 6.5C,D). Increasing chromatin condensation is seen and irregular masses of chromatin are formed. Nucleoli are not visible. The nucleus is smaller (7 to 9 µm) as is the cell as a whole (12 to 15 µm).
When the cytoplasm possesses almost its full complement of hemoglobin, the cell is termed an orthochromatic erythroblast (Figs. 6.4D and 6.5E). It is the smallest of the nucleated erythrocyte precursors (8 to 12 µm in diameter). At this stage, the nucleus undergoes pyknotic degeneration, the chromatin becomes greatly condensed, and the nucleus shrinks. It may assume various bizarre forms such as buds, rosettes, and clover leaves prior to extrusion (Fig. 6.6).
After the nucleus is extruded, the cell is known as a reticulocyte. These cells are larger than mature erythrocytes, perhaps 20% greater in volume.34 They retain certain cytoplasmic organelles, such as ribosomes, mitochondria, and the Golgi complex (Fig. 6.6C,D), and have special staining characteristics. Methyl alcohol or similar fixative agents used in staining cause precipitation of the ribosomal RNA. Such cells may thus appear uniformly blue or gray (diffuse basophilia), or basophilic shades may be intermingled with pink-staining areas (polychromatophilia or polychromasia). Certain supravital staining techniques (see Chapter 1) cause the ribosomal RNA to precipitate or aggregate into a network of strands or clumps termed reticulum; for example, cresyl blue agglutinates the ribosomes. As the reticulocyte matures, the various organelles decrease in number. Usually the mitochondria disappear first and the ribosomes last. “Autophagic vacuoles” (secondary lysosomes) containing degenerated organelles may be seen. The shape of the reticulocyte, as revealed by the scanning electron microscope, differs from that of the mature erythrocyte. Only in the late stages of maturation does the bilaterally indented disc shape of the mature red cell appear.
Flow Cytometric Analysis of Erythroid Precursors
It is now possible to broadly distinguish populations of murine or human erythroid precursors by analyzing patterns of antibodies binding to cell surface antigens. Analyses of murine hematopoietic tissues (bone marrow, spleen,31 or fetal liver35) demonstrate that the TfR (CD71) is expressed at very high levels by the early erythroid precursors, proerythroblasts, and early basophilic erythroblasts (for iron uptake for high-level heme synthesis) and that CD71 expression then decreases with erythroid maturation (and decreasing heme synthesis). On the other hand, Ter-119 (an antibody that recognizes an antigen associated with the predominant mature red cell membrane glycoprotein, glycophorin A) is expressed at intermediate levels in proerythroblasts and subsequently at high levels in more differentiated precursors.31 Thus, double immunostaining for these antigens (while excluding anucleate red cells) results in four cell populations, CD71highTer119med, CD71highTer119high, CD71medTer119high, and CD71lowTer119high corresponding broadly to proerythroblasts, basophilic, polychromatophilic, and orthochromatic erythroblasts, respectively. Similar immunostaining has also been examined during differentiation of human erythroid precursors; however, TfR expression appears to be more variable. Recent studies of human proerythroblast maturation indicate that the combination of CD36 (a high-affinity scavenger receptor36) and CD71 expression can be reliably used to discern basophilic, polychromatophilic, and orthochromatic erythroblasts by flow cytometry.37
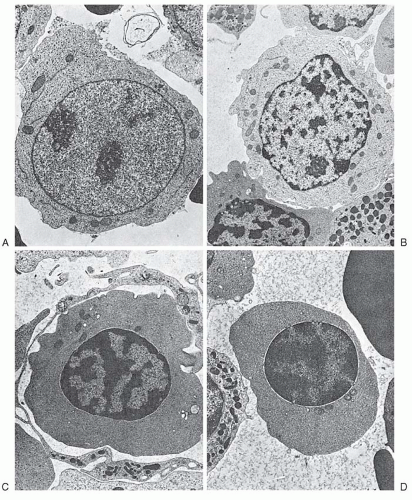 FIGURE 6.4. Maturation of erythroblasts as seen with transmission electron microscopy. Proerythroblast (A), basophilic erythroblast (B), polychromatophilic erythroblast (C), and orthochromatic erythroblast (D). (Courtesy of Dr. Carl Kjeldsberg.) |
Proliferation and Maturation of the Erythron
Within the erythron, cellular maturation and proliferation proceed simultaneously. Although BFU-E progenitors have limited self-renewal capacity, CFU-E and the erythrocyte precursors are functionally destined to mature; thus, they are incapable of self-maintenance. In response to acute demands, such as hemorrhage and hemolysis, maintenance of the erythron occurs primarily through the action of Epo promoting both progenitor proliferation (in part through increasing the CFU-E pool by reducing apoptosis19) and accelerating terminal maturation. As discussed, a majority of CFU-E progenitors, however, are already in cycle and can undergo at most three to five divisions with maximal Epo stimulation, thus limiting the erythron response. With greater or more chronic demands there appears to be an increase in BFU-E self-renewal divisions to further increase the size of the CFU-E pool.38 Obviously even greater requirements, for example, during recovery from bone marrow irradiation, necessitate input from the stem cell compartment (see Chapter 5). When severe anemia is present from birth, for example, in patients with thalassemia major, a congenital hemolytic anemia (Chapter 38), there is, in addition to maximal expansion of the various erythroid progenitor and precursor compartments, expansion of the sites of erythropoiesis from the axial bones (vertebra, pelvis, clavicles, ribs, and sternum) to other sites, potentially including the femurs, humeri, skull, spleen, liver, and even thymus.
A scheme of the proliferation of the erythron and its various stages of development is presented in Figure 6.1. It takes approximately 12 to 15 days for a cell at the BFU-E stage to mature into erythroblasts. Within 6 to 8 days, a BFU-E proliferates and differentiates into a CFU-E, which needs another 5 to 7 days to proliferate and develop into basophilic erythroblasts: a period during which the CFU-E undergoes three to five successive divisions. Probably, three to five cell divisions also occur during the maturation of erythroid precursors.39 Thus, 8 to 32 mature red cells are derived from each proerythroblast. Cell division ceases at the stage of polychromatophilic erythroblasts. Orthochromatic erythroblasts cannot synthesize DNA and, therefore, cannot divide. Two events may decrease the theoretic yield of cells. One of these is the death of erythrocytes before or shortly after release from the marrow (ineffective erythropoiesis) (Chapter 22). The second is a skipped cell division, a phenomenon that may occur with increased Epo stimulation and that results in a large hemoglobin-poor cell (Chapter 22). These events occur only to a limited extent in normal subjects but occur much more frequently under pathologic circumstances.
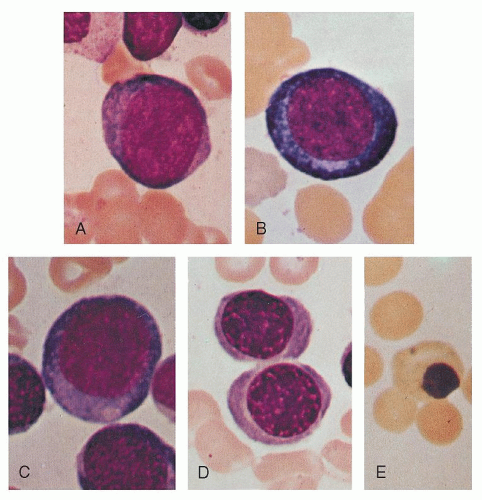 FIGURE 6.5. Erythroblasts. Proerythroblast (A); basophilic erythroblast (B); early (C) and late (D) polychromatophilic erythroblasts; orthochromatic erythroblast with stippling (E). Magnification ×1,000; Wright stain. |
The biochemical events that occur in stem cell progeny during commitment to erythroid differentiation are incompletely understood. The same holds true for the committed early erythroid progenitor BFU-E. This cell is IL-3 dependent and expresses only small numbers of EpoR.19 Within 72 hours in culture, these cells become fully dependent on Epo (“mature” BFU-E) and, in its presence, proliferate and differentiate into CFU-E progenitors.12,19 With Epo stimulation there is selective up-regulation of transcription factors, including GATA1,40 KLF1 (previously called EKLF41), and NFE2.42 GATA1 interacts with SCL/Tal1 (with LMO2, LDB1, and E2A), or with KLF1 and others in multiprotein complexes that associate with and activate (or, for example, with FOG1 and Gfi-1b and repress) erythroid genes.38,43,44,45
At this (CFU-E) stage, a number of differentiation events can be detected. From studies in murine erythroid cells, it has been established that Epo induces an increase in mRNA synthesis and that this is closely followed by the induction of murine globin gene transcription.46 Other biochemical events associated with terminal erythroid differentiation include increased uptake of calcium and glucose, synthesis of TfRs, increased iron uptake, hemoglobin synthesis, and the appearance of erythrocyte membrane proteins (e.g., bands 3 and 4.1).47,48,49 and 50 It appears that there is a GATA1-dependent phase of differentiation, controlling EpoR, antiapoptotic genes, and alpha (α-) globin gene expression that is succeeded by a KLF1-dependent phase.44,51 KLF1 appears to regulate the expression of genes essential for many key aspects of terminal erythroid differentiation including those encoding for major cytoskeletal and cell membrane proteins (ankyrin, band 3, band 4.1, dematin, and glycophorins A and C), iron transport proteins (TfR at the cell membrane and mitoferrin-1 [Mfrn1] in mitochondria52), heme synthesis enzymes, α– and beta (β)-globin chains, and α-hemoglobin stabilizing protein (AHSP, which stabilizes α-globin chains and increases their affinity for β-globin chains41,51,53). Hemoglobin synthesis continues as the cell matures into a basophilic erythroblast, and, at the polychromatophilic erythroblast stage, enough hemoglobin has accumulated in the cytoplasm to give the cell the mild acidophilic reaction that is detected by Romanowsky stains. Hemoglobin synthesis continues through the orthochromatic stage and persists at a very low rate in the reticulocyte after enucleation. Mature red cells, being devoid of ribosomes, are unable to synthesize hemoglobin.
As previously noted, morphologic evidence of decreasing erythroid precursor nuclear activity (heterochromatin formation) can be seen as early as the basophilic erythroblast stage. By the orthochromatic stage, the nucleus is completely inactive, unable to synthesize either DNA or RNA. The factors leading to cessation of nuclear activity are not fully understood, but it has been suggested that it is related to the intracellular hemoglobin concentration.54 Hemoglobin is found within the nucleus, possibly gaining entrance through pores in the nuclear membrane.54,55 and 56 After reaching a critical concentration,54 nuclear hemoglobin may react with nucleohistones, thereby bringing about chromosomal inactivation and nuclear condensation. According to this hypothesis, the number of cell divisions and the ultimate erythrocyte size are related to the rate of hemoglobin synthesis. For example, microcytic cells are produced in iron deficiency because it takes longer to reach the critical hemoglobin concentration and the generation time is unaffected; hence, more cell divisions occur before nuclear inactivation, and the resulting cell is small. In contrast, the macrocytes observed when erythropoiesis is stimulated may be the end results of an Epo-induced acceleration of hemoglobin synthesis, which in turn leads to an earlier onset of nuclear degeneration and a reduced number of cell divisions. Consistent with this hypothesis is the observation that the mean corpuscular hemoglobin concentration is relatively constant in a variety of mammalian species, even though erythrocyte size varies greatly.57 Recent studies indicate that deacetylation of nucleohistones is critical for formation of heterochromatin and nuclear condensation, and inhibition of deacetylation (by, e.g., HDAC inhibition or ectopic expression of the histone acetyl transferase Gcn5) impedes chromatin condensation and enucleation during terminal erythroid differentiation.58
After the nucleus degenerates, it is extruded from the cell.59 This process, as observed in living erythroblasts by phase contrast microscopy,60 is completed in 5 to 60 minutes. During the extrusion process, mitochondria and cytoplasmic vesicles accumulate near the nuclear border.33,61 The role of these structures in nuclear extrusion is not entirely clear, but supravital staining with Janus green B, a mitochondrial toxin, inhibits enucleation.59 The extruded nucleus carries with it a rim of cytoplasm, including ribosomes, hemoglobin, and occasional mitochondria.
Enucleation is a process similar to cytokinesis during asymmetric cell division and does not seem to depend on either the presence of extracellular matrix proteins or accessory cells.62 However, the rate of enucleation of murine erythroleukemia (MEL) cells is increased when cultured in fibronectin-coated tissue culture dishes.63 Among the various cytoskeletal proteins, filamentous actin plays an important role in the process of enucleation, accumulating between the extruding nucleus and the incipient reticulocyte (“cortical actin ring”). Supporting the major role of filamentous actin in the process of enucleation is the fact that low concentrations of cytochalasin D cause complete inhibition of enucleation.62 A Rac GTPase that activates mDia2, a formin involved in nucleation of actin filaments, is absolutely required for formation of the actin ring and enucleation.64 Colchicine, which disrupts microtubule formation also impairs enucleation.65 Interestingly, none of the major erythroid cytoskeletal proteins are found in the region of the plasma membrane that surrounds the extruded nucleus, suggesting degradation at the site of extrusion. Nurse cells are macrophages at the center of an island of erythroblasts that appear to regulate terminal erythropoiesis, supplying developmental signals and iron (and perhaps heme66) to adjacent erythroid cells.67 Erythroblast macrophage protein, expressed on erythroblasts and macrophages, appears to be important for the formation of erythroblast islands and erythroblast enucleation.68
However, as with Rac GTPase-deficient cells, EMP-deficient erythroid cells have impaired differentiation which may decrease nuclear condensation and enucleation rates.68 The importance of microRNAs (miRNA)—small nonprotein-coding RNAs that each down-regulate multiple genes posttranscriptionally—in erythropoiesis is being increasingly recognized.69 Recent studies suggest a role for miR-191 in erythroid enucleation. miR-191 is normally down-regulated with erythroid differentiation; overexpression, however, represses Mxi1 and Riok3, preventing the physiologic down-regulation of Gcn5 expression, chromatin condensation, and enucleation.38
However, as with Rac GTPase-deficient cells, EMP-deficient erythroid cells have impaired differentiation which may decrease nuclear condensation and enucleation rates.68 The importance of microRNAs (miRNA)—small nonprotein-coding RNAs that each down-regulate multiple genes posttranscriptionally—in erythropoiesis is being increasingly recognized.69 Recent studies suggest a role for miR-191 in erythroid enucleation. miR-191 is normally down-regulated with erythroid differentiation; overexpression, however, represses Mxi1 and Riok3, preventing the physiologic down-regulation of Gcn5 expression, chromatin condensation, and enucleation.38
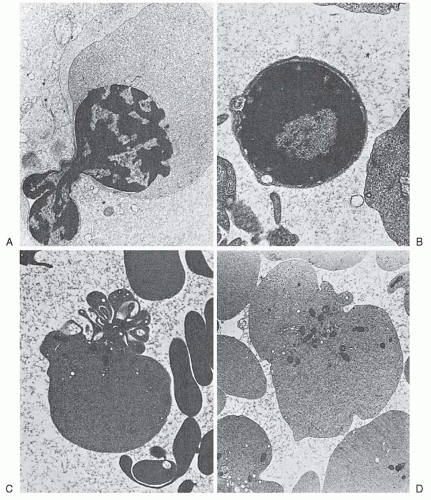 FIGURE 6.6. Formation of reticulocytes. (A) Erythroblast expelling nucleus. (B) Erythroblast nucleus after expulsion, with rim of cytoplasm. (C) Reticulocyte immediately after expulsion of nucleus. (D) Reticulocyte. (Courtesy of Dr. Carl Kjeldsberg.) |
Within the marrow, enucleation may sometimes occur as the erythroblast traverses the endothelial cell that forms the sinus wall.70 The erythroblast cytoplasm and small organelles (ribosomes and mitochondria) squeeze through endothelial cell cytoplasmic pores 1 to 4 µm in diameter, but the more rigid nucleus cannot conform to this pore size. The nucleus thus becomes caught and “pitted” from the cell. Passage through the endothelial pores is not essential to enucleation, however, because the whole process can be observed in vitro.60,62 Soon after enucleation, the nucleus is engulfed by an adjacent macrophage. The cell may remain within the marrow as a reticulocyte for several days. After release, the reticulocyte may be sequestered for 1 to 2 days in the spleen.71 Here, additional maturation may occur, and the composition of the membrane lipids may be altered. As the reticulocyte matures to an adult erythrocyte, it loses its ability to synthesize hemoglobin.72 RNA appears to be catabolized by a ribonuclease. The resulting oligonucleotides are probably further degraded by phosphodiesterases and phosphatases to pyrimidine nucleotides. A specific pyrimidine 5′-nucleotidase found in reticulocytes dephosphorylates these nucleotides, and the free pyrimidine bases can then leak out of the cell.73 If the pyrimidine 5′-nucleotidase is lacking because of hereditary deficiency73 or lead poisoning,74 RNA degradation is greatly retarded, and basophilic stippling due to retained RNA aggregates becomes prominent.
BIOSYNTHESIS OF HEMOGLOBIN
As hemoglobin accounts for approximately 90% of the dry weight of the mature red cell, the biosynthesis of hemoglobin is intimately related to erythropoiesis. As detailed in the previous section, many of the morphologic criteria used in staging the maturation of erythrocyte precursors are related to hemoglobin production and content. Furthermore, the initial events associated with the differentiation of CFU-E into erythrocyte precursors include the activation of genes relating to hemoglobin synthesis.46
Three complex metabolic pathways are required for hemoglobin synthesis, corresponding to the three structural components of hemoglobin: protein (globin), protoporphyrin, and iron. The first two of these are discussed in the pages to follow. Iron metabolism is described in Chapter 23.
Three complex metabolic pathways are required for hemoglobin synthesis, corresponding to the three structural components of hemoglobin: protein (globin), protoporphyrin, and iron. The first two of these are discussed in the pages to follow. Iron metabolism is described in Chapter 23.
Globin Synthesis
Globin Genes and the Structure of Chromatin
Distinct structural genetic loci exist for each of the polypeptide chains in hemoglobin (see also Chapter 34). Thus, there are α, β, γ, δ, and ε genes. In most human populations, the α genetic locus is duplicated, and there are four (two pairs of) identical α genes in normal subjects.75,76 There are also at least two different pairs of γ genes, one (Gγ) coding for a γ-chain with glycine at position 136 and another (Aγ) coding for a γ-chain with alanine at the same position.77 In contrast, only single (pairs of) genes code for the β-and δ-chains, respectively.
The α-gene cluster (approximately 30 kb) is located on the short arm of chromosome 16 and also contains the locus encoding for the ζ-chain,78 while the β-gene cluster (approximately 50 kb) is located on chromosome 11 and includes the genes for the Gγ-, Aγ-, δ-, and ε-globins.78,79 A schematic representation is shown in Figure 6.7.
The differentiation of erythroid progenitors to erythroblasts is accompanied by the activation of the genes involved in erythroid differentiation, including the globin genes.46,80 The active genetic regions of DNA (5% to 10% of the genetic material in erythroblasts78) make up the open portion, or euchromatin, of nuclear material, whereas unexpressed genes (the majority of genes in the nucleus) are included in the condensed, or heterochromatin, fraction. Chromatin is described as a nucleoprotein that contains, packages, and provides an instructive scaffold for nuclear DNA.81 Thus modulation of the expression of genes, including the globin genes may be imposed by the chromatin structure, which includes not only strands of DNA, but also histone and nonhistone proteins.
Transcription, Messenger RNA Processing, and Translation
Globin mRNA, like most eukaryotic mRNAs, is synthesized in a precursor form that is two to three times as long as the molecule that ultimately serves as the template for protein synthesis.75,78,80 These precursor molecules, heterogeneous nuclear RNA, undergo “processing” to be converted into the final mRNA.82 Posttranscriptional processing includes “capping” at the 5′ end of the molecule, polyadenylation at the 3′ end, and “splicing,” which results in removal of so-called intervening sequences or introns. Abnormal splicing of, for example, β-globin mRNA transcripts is a common cause of thalassemia.
The primary structure of mRNA can be divided into four regions: the 5′ untranslated region (which includes the cap), the translated or coding region, the 3′ untranslated region, and the polyadenosine region. The “cap” is characterized by an atypical 5′-triphosphate-5′ linkage with guanosine-5′-triphosphate (GTP) and methylation of adjacent nucleotides. This structure appears to be essential for maximal translational activity and enhances mRNA stability. The cap is followed by an untranslated region (“5-UTR”) of 36 nucleotide bases in α-globin mRNA and 53 bases in β-globin mRNA. The difference may explain the observation that β-mRNA is translated more efficiently than α-mRNA.78,80 Normally, this relatively inefficient translation of the α-chain is compensated for by an increased amount of α-mRNA. The translated sequence begins with an initiator sequence of three bases (AUG) followed by a sequence of triplet codons, each of which corresponds to an amino acid in globin, according to the genetic code. The translated sequence ends with a terminator codon (UAA), which is followed by a noncoding area and a terminal polyadenosine region, of variable length, that affects the stability or half-life of the molecule. The number of adenosine residues appears to decrease as mRNA ages83; in older reticulocytes, mRNA may contain little or no polyadenosine. Globin mRNA is quite stable, with a half-life of up to 48 hours, allowing reticulocytes to continue to produce hemoglobin after enucleation.
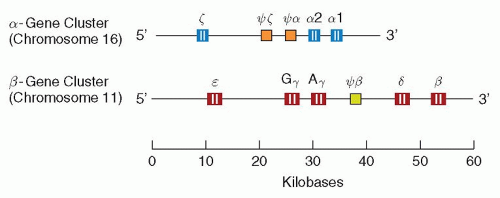 FIGURE 6.7. Organization of the human globin gene clusters on chromosomes 16 and 11. Solid areas within genes represent coding sequences; open areas represent intervening sequences. Each cluster includes pseudogenes (Ψζ, Ψα, Ψβ), which have sequence homology to functional genes but include mutations that prevent their expression. |
Regulation of Globin Synthesis
Heme is of particular importance in controlling the rate of globin synthesis.84,85 It stimulates globin synthesis in intact reticulocytes and cell-free systems, and, in its absence, polyribosomes disaggregate.86,87 and 88 The major effect of heme is exerted on the chain-initiation step in translation. In the absence of heme, an inhibitor of globin synthesis accumulates.89,90 This inhibitor, heme-regulated eIF2α kinase (HRI), acts by phosphorylating the α-subunit of an initiation factor, eIF-2 that promotes binding of tRNAmetF to ribosomes, to shut down protein synthesis.91,92 HRI, which has two heme binding sites thus serves as a sensitive intracellular heme “sensor” that closely coordinates heme availability with globin chain production to prevent the accumulation of excessive unfolded globin proteins.93 Studies of the hri knockout mouse verify the importance of this protective mechanism during high-level hemoglobin synthesis. HRI function is especially important in iron (resulting in heme) deficiency: In these circumstances, the continued globin production in hri−/− mice results in cytotoxic globin protein precipitates, causing oxidative stress, and apoptosis of late erythroid precursors.94 Interestingly, the red cells of iron deficient hri−/− mice are normocytic/hyperchromic rather than microcytic/hypochromic and have globin chain inclusions. HRI also serves to ameliorate the phenotype of β-thalassemia in mice models, minimizing the production and accumulation of α-globin aggregates, erythroid precursor apoptosis, and ineffective erythropoiesis. Recent work indicates that HRI is activated by oxidative stress (e.g., during erythropoiesis in β-thalassemia).95
In addition to its effects on globin translation, heme also exerts positive effects on globin transcription, through nuclear binding to the globin transcriptional repressor Bach1 during erythroid differentiation.96
Heme Biosynthetic Pathway
Porphyrins are heterocyclic organic rings composed of four pyrrole subunits that are usually linked by methine bridges; their conjugation to diverse divalent metal ions such as Mg2+, Co+, and Fe2+ gives rise to the “pigments of life,” i.e., chlorophyll, vitamin B12, and heme, respectively. Heme, which is a complex of ferrous iron with the tetrapyrrole protoporphyrin IX, is ubiquitous in aerobic cells and essential for cellular oxidation-reduction reactions. It serves as a critical component of hemoproteins, including cytochromes (for mitochondrial respiratory chain electron transfer and drug metabolism), oxidases (e.g., NADPH oxidase) and peroxidases, and catalases and synthases (e.g., nitric oxide synthase,
NOS), in addition to the oxygen storage and transport molecules, myoglobin and hemoglobin.97,98,99,100,101 and 102,103,104,105 and 106 The four pyrrole rings of protoporphyrin IX are designated A, B, C, and D. At the periphery of the tetrapyrrole are eight sites where side chains are located. In heme, the iron atom is inserted “like a gem”105 into the center of the tetrapyrrole. Note that heme is the ferrous iron complex of protoporphyrin IX; however, the term heme is also used in the generic sense in the literature to indicate iron protoporphyrin IX without regard to the oxidation state (valence) of the iron.
NOS), in addition to the oxygen storage and transport molecules, myoglobin and hemoglobin.97,98,99,100,101 and 102,103,104,105 and 106 The four pyrrole rings of protoporphyrin IX are designated A, B, C, and D. At the periphery of the tetrapyrrole are eight sites where side chains are located. In heme, the iron atom is inserted “like a gem”105 into the center of the tetrapyrrole. Note that heme is the ferrous iron complex of protoporphyrin IX; however, the term heme is also used in the generic sense in the literature to indicate iron protoporphyrin IX without regard to the oxidation state (valence) of the iron.
Porphyrins, by definition, are cyclicly conjugated tetrapyrroles. As such, they have a number of common properties. They are very stable, essentially flat molecules and the macrocyclic ring itself has little or no affinity for water. All porphyrins are intensely colored and they have an extremely intense absorption band at approximately 400 nm, the so-called Soret band. All porphyrins fluoresce, but fluorescence is characteristically lost when metals are bound to form metalloporphyrins. Exceptions include Mg-porphyrins and Zn-porphyrins, which fluoresce despite their metal content (Chapter 26). Of the known porphyrins, five are of importance in humans: uroporphyrin (two isomers), coproporphyrin (two isomers), and protoporphyrin (one isomer). When porphyrins are fully reduced they are called porphyrinogens. These latter compounds are colorless, do not fluoresce, cannot bind metal ions, and are extremely unstable with regard to oxidation. If uroporphyrinogen or coproporphyrinogen (intermediates in heme biosynthesis) are oxidized to their corresponding porphyrins, they can no longer function as substrates for the heme biosynthetic enzymes and must eventually be excreted in the urine and stool. Uroporphyrinogen and coproporphyrinogen can occur in four isomeric forms. Of these, only two are known to occur naturally in mammalian tissues, namely, the I and III isomer forms. Without exception, all biologically functional tetrapyrroles are derived from uroporphyrinogen III. Uroporphyrinogen I and coproporphyrinogen I are useless by-products of heme synthesis (see Chapter 26). Once formed, most uroporphyrinogen I is enzymatically decarboxylated to coproporphyrinogen I and excreted as the oxidized compound, coproporphyrin I. The difference between the type I and type III isomers is apparent on examination of the D ring. In the type I isomer, the 7 and 8 positions are occupied by acetate and propionate, respectively. In the type III isomer, the order in the D ring is reversed, and propionate and acetate are at positions 7 and 8, respectively. Note that in the case of protoporphyrin IX, the acetate at position 8 has been decarboxylated to form a methyl group (Fig. 6.8).
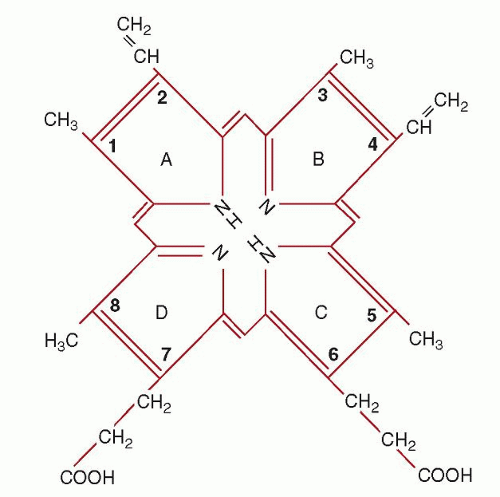 FIGURE 6.8. The chemical structure of protoporphyrin IX. |
The first of eight steps in the biosynthesis of heme (Fig. 6.9) is the condensation of glycine and succinyl coenzyme A (CoA) to yield δ-aminolevulinic acid (ALA).107,108,109 This reaction occurs in the mitochondrial matrix and is highly exergonic and essentially irreversible. The enzyme catalyzing this reaction (ALA synthase) plays a key regulatory role in the biosynthesis of heme. In the cytosol the reduced cyclic tetrapyrrole coproporphyrinogen III is formed from ALA in a series of four enzymatic reactions. Note that the product of step 2 is the monopyrrole porphobilinogen, the primary building block for all natural tetrapyrroles, including hemes, chlorophylls, and the vitamin B12 derivatives (cobalamins). How the heme synthesis intermediates are transferred from one cytosolic enzyme to the next in the pathway is at present unknown, but a macrocomplex comprising all four cytosolic enzymes, as has also been proposed for the terminal pathway enzymes, may occur.
Coproporphyrinogen III is then transported back by an unknown mechanism across the outer mitochondrial membrane into the mitochondrial intermembranous space for the three subsequent reactions required to form heme. First, propionic acid side chains at positions 2 and 4 are oxidatively decarboxylated by the enzyme coproporphyrinogen oxidase, forming protoporphyrinogen IX which is then oxidized by protoporphyrinogen IX oxidase to protoporphyrin IX. The enzymes protoporphyrinogen IX oxidase and ferrochelatase (Fech), catalyzing the penultimate and final steps of heme synthesis, are both localized at the mitochondrial inner membrane (protoporphyrinogen IX oxidase is an intermembrane space-facing protein whereas Fech is exposed to the matrix); thus, a channeling of protoporphyrinogen IX and protoporphyrin IX through a enzyme complex formed by protoporphyrinogen IX oxidase and Fech within the inner membrane has been proposed, based on biochemical studies and the crystal structure of the two enzymes.110,111 The final step, the addition of one atom of Fe2+ to protoporphyrin IX by Fech, which results in the formation of protoheme or heme b, occurs on the matrix side of the inner membrane, necessitating heme transfer, by as yet unidentified transporters, across both the inner and outer membranes again in order to reach the cytosol.
Biosynthesis of δ-Aminolevulinic Acid
The enzyme responsible for catalyzing the condensation of succinyl CoA and glycine is ALA synthase (ALAS; EC 2.3.1.37). The products of the condensation reaction are ALA, CO2, and free CoA. ALAS requires pyridoxal 5′-phosphate as a cofactor in the reaction as first suggested by nutritional studies in pigs.112 There are two forms of the enzyme, one specific for erythroid cells (ALAS2) and the other (ALAS1) present in all other tissues, especially liver. ALAS2 uniquely appears to associate with succinyl CoA synthetase in the mitochondria to promote heme synthesis.113 The genes encoding for human ALAS have been identified and cloned.114 The erythroid-specific gene is present on chromosome Xp11, whereas ALAS1 is encoded by a gene on chromosome 3p21.115,116
ALAS1 and ALAS2 differ predominantly at their amino terminal ends. The ALAS2 gene has 11 exons and extends over 22 kb. Notably, the mRNA contains a 5′ iron-response element (IRE) in exon 1. IRE are short hairpin structures that allow binding by the iron regulatory proteins 1 and 2 (IRP1 and IRP2) according to the cellular iron status, regulating the expression or stability of mRNAs encoding proteins involved in iron uptake, storage, utilization, or export117,118 (see Chapter 23). ALAS2 translation begins in exon 2, which encodes the mitochondrial targeting sequence for this nuclear-encoded gene. As also seen with ALAS1 the catalytic domain is encoded by exons 5 to 11. Mutations of ALAS2, especially of exons 5 and 9, are the most common cause of sideroblastic anemias (see Chapter 24). The promoter contains binding sites for GATA1, KLF1, and NF-E2 (although assays suggest the latter may not be functional119). However, regions further
upstream may also be important for erythroid-specific ALAS2 induction.120 In addition, analysis of intron 8 indicated the presence of DNAse I hypersensitive sites; the intron appears to contain functional binding sites for GATA1 and Sp1.121 Of relevance, recent studies identifying SLC25A38 as another frequent genetic cause of congenital sideroblastic anemias indicate it also affects either ALA synthesis or transport. Studies of yeast deficient in the yeast ortholog suggest the gene encodes a mitochondrial transporter mediating either ALA export into the cytosol or glycine import into the mitochondrial matrix, as ALA levels were low and the heme synthesis impairment was rescued by glycine or ALA supplementation.122
upstream may also be important for erythroid-specific ALAS2 induction.120 In addition, analysis of intron 8 indicated the presence of DNAse I hypersensitive sites; the intron appears to contain functional binding sites for GATA1 and Sp1.121 Of relevance, recent studies identifying SLC25A38 as another frequent genetic cause of congenital sideroblastic anemias indicate it also affects either ALA synthesis or transport. Studies of yeast deficient in the yeast ortholog suggest the gene encodes a mitochondrial transporter mediating either ALA export into the cytosol or glycine import into the mitochondrial matrix, as ALA levels were low and the heme synthesis impairment was rescued by glycine or ALA supplementation.122
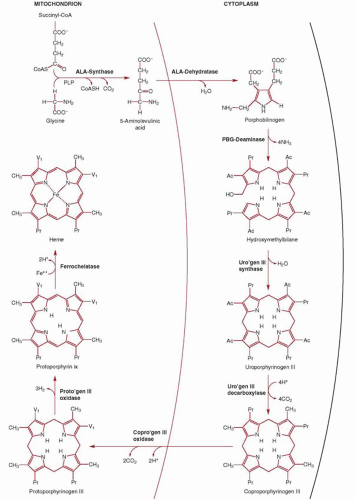 FIGURE 6.9. Heme biosynthetic pathway. Ac, acetate; ALA, δ-aminolevulinic acid; CoA, coenzyme A; CoAS, succinyl-CoA; CoASH, uncombined coenzyme A; COPRO’GEN, coproporphyrinogen; PBG, porphobilinogen; PLP, pyridoxal 5′-phosphate; Pr, propionate; PROTO’GEN, protoporphyrinogen IX (not III); URO’GEN, uroporphyrinogen; Vi, vinyl. (Modified from Bottomley SS, Eberhard Muller U. Pathophysiology of heme synthesis. Semin Hematol 1988;25:282.) |
Biosynthesis of Porphobilinogen
The monopyrrole porphobilinogen functions as a precursor of the hemes, chlorophylls, and the cobalamins.123 Because virtually all known forms of life require at least one of these classes
of tetrapyrroles, it follows that porphobilinogen is biologically ubiquitous. Porphobilinogen is formed by the condensation of two molecules of ALA and the loss of two water molecules. The enzyme that catalyzes this reaction is porphobilinogen synthase or ALA dehydratase (EC 4.2.1.24). ALA dehydratase is a soluble enzyme found in the cytosol98 and abundant in tissues such as bone marrow and liver where heme biosynthesis is active. It is also active in mature circulating erythrocytes, even though these cells are not actively synthesizing heme. Its persistence results from the enzyme’s inherent stability.
of tetrapyrroles, it follows that porphobilinogen is biologically ubiquitous. Porphobilinogen is formed by the condensation of two molecules of ALA and the loss of two water molecules. The enzyme that catalyzes this reaction is porphobilinogen synthase or ALA dehydratase (EC 4.2.1.24). ALA dehydratase is a soluble enzyme found in the cytosol98 and abundant in tissues such as bone marrow and liver where heme biosynthesis is active. It is also active in mature circulating erythrocytes, even though these cells are not actively synthesizing heme. Its persistence results from the enzyme’s inherent stability.
The mammalian enzyme is an octamer of 31 kDa subunits containing zinc atoms required for stability and activity.124 The enzyme is inhibited by heavy metals, particularly lead, which displaces the catalytically active zinc ions and thus enzyme activity can serve as an index of environmental pollution by lead.125,126 Free sulfhydryl groups (-SH) are also essential for activity,127,128 and these -SH groups seem to be protected by the zinc. Three isoforms of ALA dehydratase have been reported and the gene identified (on chromosome 9q34) and cloned.129,130 The gene consists of 12 exons, with two promoters generating different transcripts (but encoding the same protein) in erythroid and nonerythroid tissues. The erythroid promoter contains GATA1-binding and CACCC sites.
Biosynthesis of Uroporphyrinogens I and III
Porphobilinogen is a rather unstable, chemically reactive molecule. Within a few hours, a solution of porphobilinogen exposed to air and light develops a deep orange-red color. The color results from the formation of porphobilin, a poorly defined mixture of mono-, di-, and tripyrrolic oxidation products.98 This phenomenon can be observed in the urine of patients with acute intermittent porphyria, who excrete large quantities of porphobilinogen (see Chapter 30).
If instead porphobilinogen is incubated in solution at an acid pH then nonenzymic condensation or cyclization occurs, forming the tetrapyrrole macrocycle uroporphyrinogen.98 All four possible isomers of uroporphyrinogen are formed under these conditions. The reaction is often referred to as a “head-to-tail” condensation because of the apparent orientation of the precursor molecules. The penultimate reaction product apparently is an open-chain (linear) tetrapyrrole called hydroxymethylbiliane (HMB). On loss of the amino group attached to ring A, the -CH2+ can attack the free α-position on the D-ring pyrrole, thus forming the macrocycle.124
In vivo at neutral pH, these reactions are catalyzed by the cytosolic enzyme HMB synthase, formerly known as uroporphyrinogen I synthase or porphobilinogen deaminase (EC 2.5.1.61). In the cytosol HMB synthase works in concert with a second enzyme, uroporphyrinogen III synthase131 (EC 4.2.1.75), to form uroporphyrinogen III, an asymmetric cyclic tetrapyrrole that serves as the common precursor of all known functional tetrapyrroles. Uroporphyrinogen III synthase is a very fast enzyme, outcompeting the uncatalyzed reaction of HMB that results in uroporphyrinogen I (above). HMB synthase is present in humans in two tissue-specific isoenzymes, an erythroid tissue-specific and a shorter nonerythroid form, both of which are products of a single gene located on chromosome 11q23.98,124,132,133 and 134
The molecular mechanism by which uroporphyrinogen III synthase effects the “turning around” of the D ring has been studied intensively.98,135,136 The molecular mechanism involved in this reaction has been clarified by nuclear magnetic resonance spectroscopy.123,135,136 and 137 HMB synthase first catalyzes the head-totail condensation of four porphobilinogen molecules, yielding the aminomethyl tetrapyrrole. The tetrapyrrole is then deaminated, yielding a macrocycle that has been termed preuroporphyrinogen,137 which is released from the surface of HMB synthase and serves as the substrate for uroporphyrinogen III synthase.137 This enzyme opens the bond linking the methylene bridge carbon to the D ring, allowing the D ring to rotate 180° about the carbon-nitrogen bond linking rings A and D. A new carbon-carbon bond is formed linking rings C and D (1,3-sigmatropic shift). The carbon-nitrogen bond then opens, and a new carbon-carbon bond is formed linking rings A and D (1,5-sigmatropic shift) and yielding uroporphyrinogen III.137
The gene for uroporphyrinogen III synthase, located on 10q25.3, has alternative promoters in nonerythroid tissues (with transcripts starting in exon 1) and erythroid tissues (transcripts begin in exon 2). As there is no translation initiation site in exon 1, however, identical transcripts are generated. The intron between exons 1 and 2 contains multiple putative GATA1 binding sites. Mutations of the uroporphyrinogen III synthase gene are associated with congenital erythropoietic porphyria (Chapter 26).
Biosynthesis of Coproporphyrinogen III
The formation of coproporphyrinogen III is accomplished by the enzymic decarboxylation of the four acetic acid side chains of uroporphyrinogen III,99 a reaction catalyzed by the cytosolic enzyme, uroporphyrinogen decarboxylase (EC 4.1.1.37). The decarboxylation proceeds in a clockwise fashion, starting with the acetic acid on the D ring of uroporphyrinogen III and continuing with the successive decarboxylations of the acetic acid residues on rings A, B, and C.138 The enzyme exists as a dimer and it was proposed that the two catalytic centers interact functionally, perhaps shuttling reaction intermediates between subunits.139 Recent mutational studies, however, indicate that shuttling is unlikely: Dimerization mediates a more active enzyme than the monomer but without any change in the reaction products.140
The gene, located on chromosome 1p34, consists of 10 exons spread over 3 kb. Mutations of the gene encoding uroporphyrinogen decarboxylase are associated with porphyria cutanea tarda (PCT), the most common human porphyria. The structural consequences of some of these mutations have been described.141 The genetics of the disease are complex, however, with some variants without evidence of gene mutations (sporadic PCT type I; see Chapter 30 for details). A cytosolic competitive inhibitor of the enzyme, porphomethene, has been described in a murine model of PCT as a potential explanation for PCT patients with normal levels of the protein but reduced hepatic enzyme activity.142
Biosynthesis of Protoporphyrinogen IX
The formation of protoporphyrinogen IX from coproporphyrinogen III is catalyzed by the enzyme coproporphyrinogen oxidase (EC 1.3.3.3). The exact location of coproporphyrinogen oxidase in the mitochondria is unknown, with studies suggesting it is present in the intermembranous space and/or loosely associated with the inner surface of the outer membrane.143,144 It may instead form a macrocomplex with protoporphyrinogen IX oxidase and Fech to allow funneling of the substrates from the cytosol into the matrix111 (reaction products after uroporphyrinogen III being poorly soluble in aqueous solutions). The enzyme, which has a long mitochondrial targeting sequence, functions as a dimer and has an absolute requirement for molecular oxygen.143,145,146 and 147 It sequentially and oxidatively decarboxylates the propionic acid side chains in rings A and B (but not C or D) of coproporphyrinogen III to form vinyl groups.148 The transporter mediating influx of cytosolic coproporphyrinogen III into the intermembranous space is unknown.
The gene encoding coproporphyrinogen oxidase is located at 3q12, contains seven exons and spans 14 kb. Studies of the murine promoter indicate that SP1, GATA1 binding sites, and a novel transcription factor-binding sequence termed CPRE are important for induction of the enzyme during erythroid
differentiation of MEL cells.149 Mutations of the gene are associated with hereditary coproporphyria and, more rarely, with erythropoietic harderoporphyria (see Chapter 30).
differentiation of MEL cells.149 Mutations of the gene are associated with hereditary coproporphyria and, more rarely, with erythropoietic harderoporphyria (see Chapter 30).
Biosynthesis of Protoporphyrin IX
The product of the coproporphyrinogen III oxidase reaction is protoporphyrinogen IX. To serve as a substrate for the final enzyme in the pathway (ferrochelatase), protoporphyrinogen IX must first be oxidized to protoporphyrin IX. Although protoporphyrinogen IX is easily oxidized nonenzymatically to protoporphyrin in vitro (or in the cytosol), an enzyme is required to catalyze this reaction in vivo. A membrane-associated, mitochondrial, oxidizing dimeric enzyme—protoporphyrinogen IX oxidase (EC 1.3.3.4)— has been demonstrated in mammalian cells, including rat liver, human fibroblasts, reticulocytes, and leukocytes.150 The crystal structure of the tobacco plant mitochondrial enzyme has been described.111 The protein lacks a typical mitochondrial targeting leader sequence and is targeted by just 17 residues in the amino terminus.145 Flavin adenine dinucleotide (FAD) serves as an essential cofactor and molecular oxygen is utilized to terminally accept the electrons.144
The gene is present on chromosome 1q23.3 and consists of 13 exons extending over 4.2 kb. The promoters of the murine and human genes have been characterized151,152; both contain SP1 and GATA1 binding sites that may be of importance during erythroid differentiation. Mutations of the gene are associated with variegate porphyria (see Chapter 30).
Biosynthesis of Heme
The insertion of ferrous iron into protoporphyrin IX to form heme is catalyzed by the enzyme ferrochelatase within the mitochondrial matrix. Ferrochelatase (EC 4.99.1.1) is the best characterized of the heme biosynthesis enzymes.153 The enzyme, which functions as a dimer appears to be tightly bound to or is an integral part of the inner mitochondrial membrane,154 likely complexed with protoporphyrinogen IX oxidase dimers on the opposite side of the membrane.144 Insertion of the iron appears to involve distortion of the planar porphyrin by ferrochelatase.153 No cofactors are required for activity. Although the in vivo substrates are ferrous iron and protoporphyrin, in vitro, the enzyme can also catalyze incorporation of several metals (iron, cobalt, and zinc) into several dicarboxylic porphyrins (protoporphyrin, mesoporphyrin, and deuteroporphyrin).98
Studies in cell lines demonstrate that ferrochelatase interacts with Mfrn1, a mitochondrial inner membrane importer of iron up-regulated during erythroid differentiation.155 In elegant studies it has been shown that Mfrn1 is stabilized by an inner membrane ATP-binding cassette (ABC) transporter known as ABC-me or ABCB10,156 allowing high-efficiency iron uptake into mitochondria for optimal heme synthesis.157 Further studies show that ferrochelatase, Mfrn1, and ABCB10 are co-induced during MEL cell erythroid differentiation and that Mfrn1 and ABCB10 interact with ferrochelatase.155 The gene encoding ABCB10 was originally identified in a screen of mouse genes up-regulated by GATA1 and expression is regulated by heme.156 The potential substrate of the ABCB10 transporter has not been identified, but heme, which needs to be transferred from the matrix across two mitochondrial membranes into the cytosol, is a candidate.
Apart from its use for heme synthesis, mitochondrial iron is also required for mitochondrial Fe-S cluster biogenesis. Fe-S clusters are modular protein co-factors consisting of iron and sulfur, usually linked by bonds joining the cysteine sulfur atoms of a polypeptide (“scaffold”) protein to iron atoms of the cluster. They function, for example, as part of enzyme catalytic centers (e.g., aconitase, succinate dehydrogenase).158 Ferrochelatase contains a NO-sensitive Fe-S cluster that is attached at the C-terminus.153 Although the cluster is not required for catalytic function or as a supply of ferrous iron, the enzyme is sensitive to the availability of Fe-S clusters. For example, when Fe-S cluster synthesis is impaired in MEL cells, due to deficiency of the scaffold proteins or iron required for Fe-S cluster biosynthesis, then apo-Fech is rapidly degraded in mitochondria, indicating a direct link between biosynthesis of Fe-S clusters and heme.159
The gene encoding ferrochelatase is comprised of 11 exons spread out across 45 kb on chromosome 18q21.3.160 The promoter region has been examined in detail using in vitro and in vivo studies.161,162 and 163 Sp1, NF-E2, and GATA1 elements have been identified in the promoter region and a fragment of the promoter containing these binding sites allows expression in hematopoietic cells derived from transgenic embryonic mouse cells where a single copy of the reporter construct was inserted. In vivo erythroid specificity is mediated by NF-E2 elements ˜300 bp upstream of the transcriptional start site (-275 bp) along with additional erythroid-specific elements that lie between -375 bp and -1,100 bp upstream from the start site.161 In in vitro assays in K562 cells, the Kruppel-like transcription factor KLF-13 activates the promoters for porphobilinogen deaminase, δ-aminolevulinate synthase, and ferrochelatase genes.164 Mutations of the ferrochelatase gene are associated with erythropoietic porphyria (see Chapter 26).
Regulation of the Heme Biosynthetic Pathway
δ-Aminolevulinic Acid Synthase
The regulation of a biosynthetic pathway is generally effected at the first enzymatic reaction synthesizing a precursor compound committed to ultimate incorporation into the final product.165 Frequently, such reactions are strongly exergonic and essentially irreversible. These generalizations hold true for the heme biosynthetic pathway. Control of the pathway is exerted primarily through the enzyme catalyzing the first committed and rate-limiting step, ALA synthase. However, what is also apparent is that the regulation of the ubiquitous enzyme ALAS1 differs markedly from that of the erythroid-specific enzyme ALAS2.105,107
Nonerythroid ALAS1 Regulation
About 15% of the daily production of heme is generated in the liver by ALAS1 for cytochromes and enzymes. The amount of ALAS1 is regulated by induction and repression of enzyme synthesis127 and may increase by a factor as great as 300-fold.166 The enzyme has a short half-life, allowing a rapid response to changes in the demand for heme and, thus, ALA.167 The enzyme may be induced by a number of chemicals, drugs, and nonglucocorticoid steroids.127 Heme plays a critical central role in ALAS1 regulation, repressing transcription,168 decreasing the half-life of the mRNA, and, through binding to heme regulatory motifs (HRMs) in the 5′ end of the protein, translocation of the enzyme into the mitochondrial matrix.169 When the amount of intracellular heme is high, ALAS1 synthesis is repressed; when the amount of heme is low, synthesis is induced. Thus, agents that interfere with heme synthesis can induce ALAS1, and agents that induce the synthesis of hemoproteins (e.g., induction of cytochrome p450 enzymes by barbiturates), potentially depleting a putative pool of “free” or “uncommitted” heme, can produce a similar effect.170,171 Agents that exert these effects on ALAS1 synthesis induction are clinically important, as they may precipitate acute attacks in patients with acute intermittent porphyria and related disorders of porphyrin metabolism (Chapter 26).
Early studies indicated that ALAS1 synthesis is induced during fasting, which can precipitate porphyria attacks, and that nutritional supplements (e.g., glucose loading) may ameliorate
these acute episodes. Handschin et al. recently demonstrated that this occurs because ALAS1 is induced by the concerted actions of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and the transcription factors FOXO1 and nuclear respiratory factor-1 (NRF-1) on the ALAS1 promoter.172 PGC-1α, which co-activates nuclear receptors or transcription factors to regulate mitochondrial biogenesis and oxidative phosphorylation, is induced by low glucose levels or glucagon and repressed by high glucose or insulin.173 Presumably ALAS1-mediated heme synthesis is required for de novo respiratory cytochrome synthesis with mitochondrial biogenesis as a response to decreased cellular ATP levels. In contrast, regulation of ALAS2 in erythroid cells is coordinated with heme synthesis, iron assimilation, and globin synthesis.
these acute episodes. Handschin et al. recently demonstrated that this occurs because ALAS1 is induced by the concerted actions of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and the transcription factors FOXO1 and nuclear respiratory factor-1 (NRF-1) on the ALAS1 promoter.172 PGC-1α, which co-activates nuclear receptors or transcription factors to regulate mitochondrial biogenesis and oxidative phosphorylation, is induced by low glucose levels or glucagon and repressed by high glucose or insulin.173 Presumably ALAS1-mediated heme synthesis is required for de novo respiratory cytochrome synthesis with mitochondrial biogenesis as a response to decreased cellular ATP levels. In contrast, regulation of ALAS2 in erythroid cells is coordinated with heme synthesis, iron assimilation, and globin synthesis.
Erythroid ALAS2 Regulation
With erythroid differentiation there is coordination of cellular iron assimilation, heme, and globin synthesis to safely allow maximal hemoglobin synthesis within a short timespan without the buildup of individual, potentially cytotoxic components (see below). Regulation of erythroid heme involves the induction of the enzymes of the heme biosynthetic pathway and their regulation once induced,105,107 regulation of iron uptake and its delivery to ferrochelatase in the mitochondria, and the regulated export of the newly formed heme from the mitochondria to the cytosol to bind to globin chains.
Initial studies suggested that the heme biosynthesis pathway enzymes were sequentially induced with erythroid differentiation (from ALAS2 to ferrochelatase174); however, subsequent studies in MEL cells152 and maturing populations of human erythroblasts175 indicate early up-regulation of ferrochelatase with ALAS2, with synthesis of mRNAs for the terminal three pathway enzymes up-regulated within 12 hours of erythroid induction of MEL cells with dimethylsulfoxide (DMSO). In contrast to the repressive effects of heme on ALAS1 in hepatocytes, in erythroid cells intracellular heme appears to be necessary for induction of the biosynthetic pathway enzymes, perhaps through up-regulation of the erythroid transcription factor NF-E2,176 and/or inhibition of the transcriptional repressor Bach1.96 For example, studies of ALAS2-deficient MEL cells following their “erythroid” induction with DMSO demonstrate a lack of erythroid differentiation (as assessed by a lack of up-regulation of mRNAs for ALA dehydratase, porphobilinogen deaminase, ferrochelatase, and β-globin) that is at least partially reversed by addition of heme to DMSO, in keeping with studies by others of ALAS2-deficient cell lines.177
As discussed above, unlike ALAS1, there is an IRE in the 5′UTR of the ALAS2 gene. Therefore depletion of cytosolic iron (believed to exist in a putative labile iron pool) in erythroid precursors should result in binding by IRP1 and IRP2 to the IRE of ALAS2 transcripts, preventing translation. Thus, it is the supply of iron to the erythroid precursor that ultimately controls heme synthesis. To allow maximal heme synthesis—which requires ˜20 mg of iron for the 20 g of erythrocytes generated in adult humans every day—iron is delivered to the bone marrow in the form of ferric-transferrin (Fe-Tf) that is rapidly bound by TfR1 present in large numbers on the cell surface of erythroid precursors (up to 106 receptors per cell178). In addition, as noted above, the mitochondrial iron importer MFN1 becomes stabilized in a macrocomplex with ferrochelatase and ABCB10 following up-regulation of ABCB10 with erythroid differentiation, thus facilitating the transfer of iron across the mitochondrial inner membrane to ferrochelatase. By necessity, there must also be up-regulation of an unidentified heme transporter to export heme from the mitochondria into the cytosol. As a feedback mechanism “uncommitted” or “free” heme appears to inhibit either Fe-Tf-TfR endocytosis or iron release from Tf to prevent unnecessary iron uptake.105,179 Like ALAS1, ALAS2 also has HRMs located in the 5′ end of the preALAS2 protein and, in vitro, micromolar quantities of heme inhibit translocation of ALAS2 into isolated mitochondria; however, whether excess uncommitted heme also impedes ALAS2 translocation in erythroid precursors is unknown.169
ALAS2 and ferritin transcripts both contain an IRE in their 5′-UTR and their translation is thus susceptible to low cytosolic iron levels; on the other hand, TfR1 contains five IRE modules in its long 3′-UTR and the mRNA should be stabilized (predominantly by IRP2180) under the same conditions. However, the standard posttranscriptional regulatory model involving cytosolic iron levels controlling iron transport, utilization, and storage via the IRE/IRP system (see Chapter 23) appears to become uncoupled in differentiating erythroid cells: Here ALAS2 is translated yet ferritin translation blocked, whereas high expression of TfR1 persists despite high Fe-Tf delivery. Recent analyses of mass cultures of erythroid progenitors indicate that, with differentiation, these cells behave as if a “low cytosolic iron level” condition exists.181 This is in keeping with the “kiss and run” hypothesis of iron delivery proposed by Ponka and co-workers,182 whereby iron released from Fe-Tf-TfR complexes in the endosome bypasses the cytosol to be delivered to the mitochondria by direct contact between these two organelles. An alternative theory is that MFN1 functions as a highly efficient mitochondrial iron importer driving cytosolic iron transfer across inner mitochondrial membranes for heme or Fe/S synthesis. In addition, it is suggested that the presence of large numbers of ALAS2 transcripts may overwhelm the IRE/IRP system.181
Critical Balance between Iron Assimilation, Heme, and Globin Synthesis
In closing this section, note should be made of a number of recently described mechanisms to protect erythroid precursors from the results of any imbalance among iron assimilation, heme, and globin synthesis. Although each component is essential for hemoglobin synthesis, individually they are all potentially cytotoxic and it is therefore crucial that the α– and β-globin chains and heme are produced in the 2:2:4 ratio necessary to form the stable complex of α2β2 and 4 heme molecules that comprise hemoglobin A. The toxicity of free iron is well known, related to its intrinsic ability to generate highly reactive hydroxyl radicals from hydrogen peroxide in the Fenton reaction, whereas “free” or uncommitted heme is lipophilic and toxic to cells, promoting lipid peroxidation and ROS production, resulting in membrane injury and ultimately cell apoptosis.106,183,184 The cytotoxicity derived from an imbalance in the production of α-globin and β-globin chains is best illustrated by the pathophysiology of β-thalassemia (Chapter 34), where the relative excess in α-globin production and the resultant precipitation of these globin chains triggers oxidative stress and cytotoxicity (first described in the 1960s185,186).
These protective systems include the following.
(i) The heme-regulated inhibitor of translation, HRI
As mentioned, HRI, a heme-regulated protein kinase that phosphorylates and inhibits eIF2α and thus general protein translation, serves as a sensor of intracellular heme and is important for coordinating heme and globin production. Studies of the hri knockout mouse verify the importance of this protective mechanism during high-level hemoglobin synthesis. HRI function appears to be especially important in iron (resulting in heme) deficiency: In these circumstances, the cessation of globin production seen in control mice presumably does not occur in hri−/− mice, resulting in the observed cytotoxic globin protein precipitates, ROS production and oxidative stress, and apoptosis of late erythroid precursors.94 Interestingly, the red cells of irondeficient hri−/− mice are normocytic/hyperchromic (rather than
microcytic/hypochromic, as observed in the iron-deficient control mice), have globin chain inclusions, and a more severe anemia. Similarly, mice with combined HRI and severe ferrochelatase deficiency (Fechm1Pas/m1Pas mice, another model of heme deficiency) have more severe anemia and globin chain inclusions than control Fechm1Pas/m1Pas mice. Notably, these mice have a 30-fold increase in red cell protoporphyrin IX compared with Fechm1Pas/m1Pas mice, emphasizing that HRI-mediated regulation of erythroid precursor protein synthesis also affects heme enzyme biosynthesis and the severity of the porphyria.172
microcytic/hypochromic, as observed in the iron-deficient control mice), have globin chain inclusions, and a more severe anemia. Similarly, mice with combined HRI and severe ferrochelatase deficiency (Fechm1Pas/m1Pas mice, another model of heme deficiency) have more severe anemia and globin chain inclusions than control Fechm1Pas/m1Pas mice. Notably, these mice have a 30-fold increase in red cell protoporphyrin IX compared with Fechm1Pas/m1Pas mice, emphasizing that HRI-mediated regulation of erythroid precursor protein synthesis also affects heme enzyme biosynthesis and the severity of the porphyria.172
HRI also ameliorates the phenotype of β-thalassemia in mice models, minimizing the imbalance in production between α– and β-globins, the accumulation of α-globin aggregates, apoptosis of erythroid precursors, and the resultant ineffective erythropoiesis. Recent work indicates that HRI is also activated by oxidative stress (e.g., during stress erythropoiesis or erythropoiesis in β-thalassemia).95 HRI, by activating the transcription factor ATf4, induces an antioxidant stress response that seems to be important for erythroid differentiation.
(ii) The feline leukemia virus subgroup C receptor, FLVCR
FLVCR1 is the human ortholog of the feline cell surface receptor for feline leukemia virus subgroup C (FeLV-C). The virus infects all feline hematopoietic cells, impairing feline FLVCR1 function due to binding of the receptor by viral envelope that is continuously synthesized within infected cells. Cats infected with FeLV-C develop a red cell aplasia characterized by a block in erythroid differentiation at the CFU-E/proerythroblast stage. The impairment in differentiation is also observed upon conditional deletion of flvcr1 in neonatal mice,187 who develop a severe anemia within 5 weeks of deletion187 that may be due to erythroid cell apoptosis.188 FLVCR1 functions as a mammalian cell-surface heme exporter that thus appears to protect differentiating erythroid progenitors from potential heme excesses and subsequent cytotoxicity resulting from any imbalances between heme and globin synthesis.104,188 As noted, heme synthesis by erythroid ALAS2 is not subject to transcriptional repression by heme and catabolism by heme oxygenases does not normally occur during differentiation of human erythroid progenitors189 or murine erythroid cell lines.190 It is hypothesized that heme oxygenase is not induced in order to prevent futile cycles of simultaneous erythroid heme synthesis and catabolism.188,189 The severity of the anemia in feline and murine models of FLVCR knockdown suggests that excess heme synthesis occurs frequently at the CFU-E/proerythroblast stage (likely prior to initiation of high-level globin synthesis) or that FLVCR has yet another cell function. A recent study suggests another FLVCR isoform functions to export heme from its site of synthesis in the mitochondria into the cytosol190a. Of interest, knockdown of murine flvcr1 is embryonic lethal with the embryos displaying a phenotype similar to that of patients with Diamond-Blackfan anemia (DBA), a congenital red cell aplasia.191 The most common genetic cause of DBA is haplo-insufficiency of large or small ribosomal protein subunits, impairing erythroid protein (i.e., predominantly globin) synthesis, suggesting DBA is caused by an imbalance of heme and globin synthesis.192
(iii) The α-hemoglobin stabilizing protein, AHSP
The gene encoding this small protein (102 aa) is strongly induced by GATA-1 during erythroid differentiation. AHSP primarily binds αHb (i.e., the holoprotein, α-globin-heme), stabilizing it and inhibiting its pro-oxidant properties.53 It functions as a chaperone, helping newly synthesized apo α-globin chains to fold and promoting the refolding of denatured chains, which may be particularly important in heme deficiency.193 Studies have shown that AHSP forms a heterodimer with αHb and when βHb is added to these complexes, the AHSP is displaced and tetrameric HbA (α2β2) forms, suggesting that AHSP stabilizes αHb and then passes it to βHb to help form HbA in vivo. Deletion of the gene in mice194 results in a mild hemolytic anemia with the red cells containing Heinz bodies (eosinophilic inclusions derived from denatured hemoglobin), indicating perhaps that AHSP function is critical only when there is a large imbalance in synthesis between αHb and βHb, such as occurs in β-thalassemia. In subsequent studies, interbreeding of AHSP−/− mice with β-thalassemic mice (like the Hri knockout above) was indeed shown to worsen the β-thalassemic phenotype.195
Lastly, “protein quality control mechanisms” has been proposed as a name for a number of cellular posttranslational mechanisms that serve to stabilize and aid folding of newly forming proteins (e.g., chaperones such as AHSP), or recognize misfolded proteins or protein aggregates and target them for degradation by the ubiquitin proteosome system or autophagy.196 These protein quality control mechanisms appear to be particularly important in erythroid precursors during high-level hemoglobin synthesis, and when these systems are overwhelmed—for example, in severe thalassemias—accumulation of unstable insoluble proteins and cytotoxicity occurs.197
CONTROL OF ERYTHROPOIESIS
It is evident that a well-balanced mechanism exists that maintains the erythron within “normal” limits and mediates the response to a variety of normal and abnormal situations. In broad outlines, this control system operates in the following manner. Alterations in the concentration of hemoglobin in the blood lead to changes in tissue oxygen tension within the kidney. In response to hypoxia, the kidney secretes a hormone called Epo. This hormone induces differentiation of erythroid progenitor cells, expansion of the erythroid marrow, and increased red cell production. This, in turn, leads to an increase in the size of the erythron and an increase in tissue oxygen levels. The major steps in this process are discussed in greater detail in the sections that follow.
Tissue Oxygen
Tissue oxygen tension depends on the relative rates of oxygen supply and demand. Oxygen supply is a complex function of interacting but semi-independent variables, including (a) blood flow, (b) blood hemoglobin concentration, (c) hemoglobin oxygen saturation, and (d) hemoglobin oxygen affinity. Each of these functions may be altered to compensate for a deficiency in one of the others. For example, in severe anemia, cardiac output and respiratory rate may increase, and hemoglobin oxygen affinity may be reduced through the 2,3-biphosphoglycerate effect. Conversely, in respiratory insufficiency, secondary polycythemia occurs.
Despite cardiovascular and respiratory adjustments, tissue oxygen tension decreases roughly in proportion to the degree of anemia. Conversely, induced polycythemia of moderate degree leads to normal or increased tissue oxygen tension and an increased tolerance to hypoxia. These changes occur despite the increase in blood viscosity that accompanies polycythemia, suggesting that peripheral vascular resistance decreases to compensate for increased viscosity. However, with advanced degrees of polycythemia, the increase in viscosity may be great enough to negate the advantages of increased oxygen-carrying capacity.
Tissue hypoxia is the fundamental stimulus to erythropoiesis, as first suggested by Miescher in 1893. This concept has been amply confirmed.2 However, hypoxia does not exert its effects by a direct action on the marrow, as Miescher believed, but instead induces a hormone, Epo. The nature of the tissue oxygen receptors (or oxygen sensor) has only recently been understood. These sensors are located within the kidney and Epo production can be induced by renal artery constriction or by hypoxic perfusion of the isolated kidney.
Erythropoietin
Structure of Erythropoietin
Epo is a glycoprotein hormone produced by the kidney, which functions as the major humoral regulator of red cell production. The hormone was originally purified from the urine of patients with aplastic anemia.198 It has an MW of 34 kDa as determined electrophoretically and contains 30% carbohydrate (11% sialic acid, 11% total hexose, and 8% N-acetylglucosamine).199 The potency of Epo is expressed in units, with one unit defined as the amount of Epo present in one tenth of the International Reference Preparation.200 This unit was originally defined in starved rats as the amount of Epo that produced the same erythropoietic response (increase in serum Epo level) as treatment with 5 µmol of cobalt.2 The potency of purified human urinary Epo has been determined to be 70,400 U/mg of protein.198
The Epo gene has been isolated and cloned201; the genomic locus extends over 5.4 kb on chromosome 7q22202 and contains four introns and five exons, encoding a 193-amino acid polypeptide. The protein includes a 27-amino acid signal peptide, which is cleared during Epo secretion, and a 166-amino acid peptide with an MW of 18.4 kDa.46 The C-terminal arginine is absent from the recombinant and native proteins, presumably because of posttranslational modification by a carboxypeptidase.203 Human Epo contains four cysteine residues linked by disulfide bonds, which, when reduced or alkylated, lead to significant loss of activity.199,204
Recombinant Epo (rEpo), as synthesized by mammalian cells such as a Chinese hamster ovary cell line, is highly glycosylated, and the carbohydrate structure of the recombinant hormone is similar to but distinct from the glycosylation pattern of native Epo in kidney cells.203 Glycosylation is absolutely necessary for in vivo activity of Epo, with the bulk of glycosylation occurring at a single site of N-linked carbohydrate. Asialated Epo and nonglycosylated rEpo produced in bacteria have no activity in vivo, which is at least partially attributed to rapid clearance of the hormone by the liver via hepatocyte galactose receptors.205,206
Recognition of the importance of glycosylation of Epo to its in vivo activity and half-life led to modifications of the Epo gene/protein to make a more effective pharmaceutical. For example, the gene has been modified by adding a second site of N-linked glycosylation, such that when the gene is expressed in Chinese hamster ovary cells, the amount of carbohydrate attached to the modified protein is almost doubled. This new product, called darbepoetin, has a longer in vivo half-life than rEpo, thus fewer injections per week are required for therapeutic efficacy.207
Studies on the amino acid sequences of human and murine Epo have shown a very high degree of conservation of the molecule structure in these two species.208,209 Analyses of the NMR structure of Epo and the crystal structure of Epo complexed to the extracellular component of the EpoR have been performed.210 Similar to the structure of hGH and GCSF (Chapter 5), Epo consists topologically of a bundle of α-helices (four in Epo210). Two Epo-EpoR binding sites on Epo have been identified, a high-affinity site 1 (Kd ˜ 1 nM) and low-affinity site 2 (Kd ˜ 1 µM).211 The studies are in keeping with the interaction of a single molecule of Epo with EpoR dimers.
Site and Regulation of Erythropoietin Production
More than 50 years ago Jacobson et al. established that the kidney is the major site of Epo production in adult rats.212 Humans with end-stage renal failure were also found to have low serum Epo concentrations, which were restored to normal following renal transplantation.213 The cloning of the murine Epo gene has allowed studies on the production of Epo-specific mRNA in anemic mice. Induction of anemia leads, within an hour, to the appearance of Epo-encoding mRNA in the kidney and liver of anemic mice and rats.213,214 After bleeding, the Epo mRNA in the kidney increases 500 to 1,000 times when compared with Epo mRNA levels in normal kidney, whereas the liver produces only 7% of the total Epo mRNA.215 These changes in Epo mRNA synthesis are followed by parallel changes in serum Epo concentration, as determined by radioimmunoassay, indicating that Epo production in response to anemia represents de novo synthesis rather than the release of preformed hormone.214 Murine Epo mRNA is detectable by ribonuclease protection assay at 14 days of gestation in the fetal liver and a week later in the kidneys, which assume a major role in Epo production after birth.216 In cases of paraneoplastic erythrocytosis, Epo mRNA is detected in the neoplastic cells.217,218
Specialized cells producing Epo have been identified in renal and hepatic parenchyma by in situ hybridization techniques, using radioactive probes specific for Epo mRNA.216,219,220 These rare Epo-producing cells are found in the renal parenchymal interstitium (outside the tubular basement membrane), predominantly in the inner cortex and outer medulla. The bulk of experimental evidence indicates that these are fibroblast-like type I interstitial cells.221,222 In the liver, Epo mRNA is detected in hepatocytes. The number of interstitial renal Epo-producing cells increases (approximately exponentially) in response to anemia, indicating that increased Epo production is met by an increase in the number of Epo-producing cells; presumably with worsening anemia, increased numbers of these cells become sufficiently hypoxic to trigger Epo synthesis.62 Notably, there is no detectable storage of the hormone, and increased levels of circulating Epo do not repress further Epo production. Epo and EpoR are also expressed at low levels in other tissues including the spleen, bone marrow, lung, testis, eye, and brain.223 A possible paracrine function, potentially supporting low-level erythropoiesis, has been ascribed to Epo production within bone marrow hematopoietic progenitors,224,225 and production of Epo within the CNS appears to protect EpoR-bearing neurons from ischemic damage and apoptosis.226,227
The mechanism by which hypoxia leads to Epo synthesis has been determined. A sequence located in a region flanking the 3′ end of the Epo gene is oxygen-sensitive and involved in regulation of expression.227 It has been shown, for example, that this oxygen-sensitive enhancer sequence when fused to a plasmid construct comprising the Epo promoter and a reporter gene confers to transfected cells the ability to respond to hypoxia as detected by an increase of the protein encoded by the reporter gene and studies of transgenic mice expressing defined 5′ or 3′ sequences of the human Epo gene support these findings.228 The ligand for this oxygen-sensitive enhancer was identified as a 120 kDa protein termed hypoxia-inducible factor 1 (HIF-1).228,229 This DNAbinding protein is tightly regulated by intracellular oxygen tension and serves as the physiologic regulator of Epo transcription.230
HIFs are heterodimeric helix-loop-helix transcription factors consisting of two subunits, an oxygen-labile protein, HIF-α, and a constitutively expressed β subunit, HIF-β.231 Note that three genes, HIF1A, HIF2A or EPAS1, and HIF3A, encoding different isoforms of HIF-α are present in the human genome; here HIF-α refers to HIF-1α or HIF-2α. The concentration and transcriptional activity of HIF-α increase in a geometric fashion upon exposure to hypoxia. HIF-α mRNA is constitutively expressed under normoxic conditions, but the protein is rapidly degraded via the ubiquitin proteosome complex following binding by von Hippel-Lindau protein (pVHL). The recognition of HIF-α by pVHL requires prior hydroxylation of specific HIF-α proline residues by prolyl-hydroxylase domain (PHD)-containing proteins.232,233,234 and 235 These PHDs are oxygen- and iron-dependent enzymes. Under hypoxic conditions, little or no proline hydroxylation takes place; thus, pVHL does not bind to HIF-α, which accumulates in the nucleus, heterodimerizes with HIF-β, and recruits the transcriptional coactivators p300/CREB-binding
protein, with the whole complex then binding to the Epo enhancer to positively influence Epo promoter activity and gene transcription. The recruitment of p300 to the complex can itself be inhibited by hydroxylation of asparagine-803 in HIF-α, which is catalyzed by asparaginyl-hydroxylase, another oxygen-sensitive enzyme.236 It seems that these two amino acid hydroxylases, by their dependence on normal intracellular oxygen for their function, act as the oxygen sensor in the Epo-producing interstitial cells in the kidney, and, by regulating the function of HIF-α at two distinct points,235 ultimately control Epo synthesis and production. Not surprisingly, mutations of this pathway may be associated with an increased red cell mass: Chuvash polycythemia is due to a homozygous mutation of Vhl that impairs HIF-1α degradation, resulting in a mild increase in Epo levels237 whereas mutations of the genes encoding HIF-2α or PHD2 are rarer causes of familial erythrocytosis.238
protein, with the whole complex then binding to the Epo enhancer to positively influence Epo promoter activity and gene transcription. The recruitment of p300 to the complex can itself be inhibited by hydroxylation of asparagine-803 in HIF-α, which is catalyzed by asparaginyl-hydroxylase, another oxygen-sensitive enzyme.236 It seems that these two amino acid hydroxylases, by their dependence on normal intracellular oxygen for their function, act as the oxygen sensor in the Epo-producing interstitial cells in the kidney, and, by regulating the function of HIF-α at two distinct points,235 ultimately control Epo synthesis and production. Not surprisingly, mutations of this pathway may be associated with an increased red cell mass: Chuvash polycythemia is due to a homozygous mutation of Vhl that impairs HIF-1α degradation, resulting in a mild increase in Epo levels237 whereas mutations of the genes encoding HIF-2α or PHD2 are rarer causes of familial erythrocytosis.238
In addition to Epo, a large number of other HIF target genes (e.g., glucose transporters, glycolytic enzymes, and vascular endothelial growth factors) are up-regulated during hypoxia to aid cells adapt to hypoxic conditions.239 Apart from the indirect effects of hypoxia on erythropoiesis through HIF-mediated renal Epo production, erythroid progenitor cells are also subject to direct cellular effects of hypoxia and HIF production. A recent analysis of early erythroid progenitor genes up-regulated by glucocorticoids (such as cortisol, which is released from the adrenal glands during acute anemic or hypoxic “stress erythropoiesis”38,240) found that the promoter regions of many of these erythroid cell genes also contain HIF binding sites. Furthermore, in in vitro studies, HIF synergizes with glucocorticoids to expand erythroid progenitors dramatically, perhaps by increasing BFU-E progenitor self-renewal,38 a HIF effect previously observed by others during stress erythropoiesis.241
Action of Erythropoietin
Erythropoietin Receptors
Epo binds to specific molecules on the cell surface, the EpoR. The expression of both Epo and EpoR is necessary for adult life. Deletion of either of the genes encoding for Epo or EpoR in mice results in the identical phenotype of fetal death at embryonic days E11.5 to E13.5 because of a lack of definitive erythropoiesis in the fetal liver and severe anemia.242 Arguably, the most important control point of erythropoiesis is the interaction of Epo with the receptor for Epo.38,243,244 and 245 The activation of EpoR generates an intracellular signal in immature erythroid cells that promotes survival of cells that would otherwise undergo apoptosis. Epo also appears to promote erythroblast proliferation and differentiation.
EpoR is expressed on hematopoietic cells that respond to Epo and has been identified on human12 and murine erythroid cells,216 on erythroleukemia cell lines,216 in murine fetal liver tissue rich in erythroid elements, in mouse and rat placenta,217,246 and on megakaryocytes.246,247 EpoR expression on erythroid cells is relatively low (approximately 1,000 molecules per cell) and correlates with the cell’s responsiveness to and dependence on Epo.247 EpoR is detectable by autoradiography on human BFU-E; its density increases as BFU-E matures to CFU-E.19 Erythroid cells at a stage between CFU-E and proerythroblast seem to have the highest density of EpoR, which decreases as the proerythroblast matures and eventually disappears at the stage of orthochromatic erythroblast.19,246 The receptor is not expressed on reticulocytes or red cells. The presence of EpoR on megakaryocytes246,247 and 248 explains why Epo at physiologic concentrations promotes megakaryocyte differentiation and can thus affect platelet levels. Receptors for Epo are also observed on nonhematopoietic tissues including neurons and cardiac myocytes, endothelial cells, the kidneys, and embryonic muscle. The expression of EpoR in nonerythroid tissues was not believed to be required for normal embryonic development as erythroid tissue-specific EpoR expression (under control of the GATA-1 promoter) in EpoR−/− embryonic stem cells gives rise to apparently normal mice.249 However, recent studies indicate that EpoR−/− murine embryos develop neural defects,250 defects in angiogenesis,251 and cardiac ventricular hypoplasia, which is not a result of generalized hypoxia.252 In addition, some of the adverse effects observed in patients with certain solid tumors receiving Epo have been ascribed to activation of EpoR expressed on tumor cells.253
The MEL EpoR gene encodes a 507-amino acid peptide of 62 kDa with extracellular, single-membrane-spanning and intracellular domains.254 The EpoR undergoes glycosylation and phosphorylation before being incorporated into the cell membrane as a 70 to 78 kDa protein.254 Cells transfected with this gene express both high- and low-affinity EpoR.254 The human gene is located on chromosome 19p13.2, encodes a protein of 508 aa, and is 68 to 72 kDa depending on the degree of glycosylation.255
It is now recognized that structurally EpoR is part of a large family of type I cytokine receptors, which includes receptors for IL-2 to IL-7, GM-CSF, and Tpo. Type I cytokine receptors share basic structural features and are characterized by four conserved cysteine residues and a tryptophan-serine-x-serine-tryptophan (WSXSW) motif in the extracellular domain and by conserved box1/box2 regions in the intracytoplasmic domain adjacent to the membrane. Some of these type I cytokine receptors share common subunits and thus are heterodimeric; however, the receptors for Epo, Tpo, and G-CSF consist of homodimers.256,257 Crystallographic studies confirm that, as is seen with other cytokine receptors, one molecule of Epo simultaneously binds to two EpoR.258 After binding, both Epo and EpoR are rapidly endocytosed and degraded.12,216,247,255 Although it is clear that EpoR activation by Epo can lead to formation of EpoR homodimers,259 evidence also indicates these dimers exist prior to Epo binding, and that binding shifts and stabilizes an active receptor conformation bringing the two EpoR into closer contact.260,261
EpoR signaling pathways are outlined in Figure 6.10. Tyrosine phosphorylation of the EpoR262 is the first observable event after Epo binding. Because EpoR lacks a kinase domain, a tyrosine protein kinase must therefore associate with the receptor. JAK2, a member of the Janus family of cytoplasmic tyrosine kinases, is the primary EpoR-associated kinase and binds to a conserved sequence of amino acids found in cytoplasmic domains of the EpoR263,264 (and other receptors related in sequence to EpoR). Interestingly, recent studies indicate that the activation of EpoR through binding of Epo initiates a scissorlike rotation of the EpoR dimers, separating the intracellular domains of the two receptor molecules to allow room for the associated Jak2 molecules; in addition there appears to be a self-rotation of each monomer to allow them to orient properly for transphosphorylation of Jak2.265 Deletion of the JAK2 gene in mice results in fetal death on days 12 to 13 that is associated with a severe anemia mirroring the phenotype after deletion of Epo or EpoR,266 indicating the importance of Jak2 for Epo signaling.
To summarize current research,38,243,244 and 245 one Epo molecule binds two EpoR molecules, activating JAK2 kinases associated with the juxtamembrane regions (box 1/box 2) of each receptor by physically bringing the inactive (or low-activity) kinases into close proximity during the induced rotational shift in EpoR conformation,267 such that these kinases cross-phosphorylate each other, gaining full activity. The activated kinases phosphorylate all eight conserved tyrosine residues of the EpoR cytoplasmic tail. The phosphorylated tyrosine (PY) residues then serve as docking sites for up to 20 different signaling molecules or adaptor proteins that may be phosphorylated by JAK2 to become active, and leading to various mitogenic, differentiative, and antiapoptotic responses. The signaling molecules and adaptor proteins contain either Src homology 2 (SH2) or other phosphotyrosine binding domains that mediate recognition of a PY residue in the context of specific
adjacent (EpoR) amino acids. The major pathways activated by Epo binding to EpoR include the signal transduction activator of transcription 5 (STAT5) pathway, and the phosphatidylinositol-3-kinase (PI3-kinase)/Akt and ras/raf/MAPK (mitogen-activated protein kinase) signaling cascades (Fig. 6.10).
adjacent (EpoR) amino acids. The major pathways activated by Epo binding to EpoR include the signal transduction activator of transcription 5 (STAT5) pathway, and the phosphatidylinositol-3-kinase (PI3-kinase)/Akt and ras/raf/MAPK (mitogen-activated protein kinase) signaling cascades (Fig. 6.10).
 FIGURE 6.10. Overview of the erythropoietin receptor signaling pathways. Conformational changes in EpoR dimers induced by Epo binding facilitate the activation of EpoR-associated Jak2 kinases. Jak2 kinase activation results in phosphorylation of several tyrosines in the EpoR cytoplasmic tail that then serve as docking sites for signaling or adaptor proteins containing phosphotyrosine-binding domains. The signaling proteins become phosphorylated and function in numerous downstream signaling cascades. (1) A major target of Jak2 is Stat5, which is phosphorylated, dimerizes, and travels to the nucleus where it activates genes important for cell survival and proliferation. (2) Binding of the regulatory subunit of PI3-kinase, p85, to EpoR phosphotyrosines results in phosphorylation of membrane lipids and the downstream activation of Akt. This kinase regulates cell-cycle, cell differentiation, and pro-survival pathways and modulates metabolism and protein translation. (3) The ras/raf/MEK/ERK pathway is activated with binding of the SHC/GRB2/SOS complex to EpoR phosphotyrosines. Activation of the downstream kinase ERK1/2 results in phosphorylation of ELK1, a transcription factor important for cell survival and proliferation (via c-jun and c-fos expression). Negative regulatory proteins are necessary to dampen EpoR signaling. SOCS proteins inhibit Jak2 and Stat5 activation whereas phosphatases such as SHP1 and SHIP inhibit other phosphorylationdependent pathways. Note that other EpoR pathways, such as activation of calcium-dependent isoforms of the PKC family of serine/threonine kinases, are not shown. See text for details. |
Upon binding to EpoR at PY343 (EpoR numbering here refers to the murine protein), STAT5 is phosphorylated by Jak2,268 dimerizes, and translocates to the nucleus to mediate gene transcription (e.g., of the mitogenic transcription factor c-myc, Id1,10 TfR1, and IRP-2269). Targeted deletions of the eight potential PY residues in EpoR, to dissect the Epo/EpoR/Jak2 signaling axis, have also revealed the importance of pathways downstream of Jak2.270 Although deletion of all eight residues results (surprisingly) in only mild anemia (the hematocrit in adult mice is reduced by 25% to ˜0.37),271 restoration solely of Epo/EpoR/Jak2/STAT5 signaling (i.e., by deletion of all tyrosines except the Y343 residue) results in near-wild-type EpoR activities,272 and in particular, STAT5 signaling restores the erythron capacity for proliferative, stress erythropoiesis responses. The precise role of STAT5 in EpoR signaling in steady-state erythropoiesis, however, is still unclear. STAT5 is expressed from two very similar genes, STAT5a and STAT5b. In animal studies in which both genes were deleted or “knocked out” (STAT5a/b−/− mice), a marked fetal anemia was reported with lesser but significant anemia in newborn and adult animals.273 More recent studies indicate these knockout STAT5a/b−/− animals contain hypomorphic rather than null STAT5a/b−/− alleles, expressing an N-terminally truncated phosphorylated Stat5 in erythroblasts that appears to retain some biologic STAT5 functions.274 In addition, Epo weakly activates other STATs in primary erythroid cells, such that STAT3269 and/or STAT1 activation274 may also partially compensate for STAT5, resulting in a relatively mild inhibition of murine erythropoiesis.275
A potential target of STAT5 is the Bcl-x gene, which encodes an antiapoptotic protein, Bcl-xL,, believed to be essential for Epo-dependent survival. Two recent studies of Bcl-xL-deficient animals confirmed Bcl-xL expression is required for normal erythropoiesis, but also demonstrate that it promotes the survival of mature erythroid cells that no longer depend on Epo for survival.276,277 Thus, Bcl-xL must also be regulated by factors other than Epoactivated STAT5,274 likely the interplay of GATA-1 and Gf-1B at the Bcl-x promoter.278 Epo/EpoR signaling may promote survival earlier, at the proerythroblast stage, through up-regulation of a
number of other antiapoptotic genes including Pim1, Pim3, Trib3, and Serpina 3g.255
number of other antiapoptotic genes including Pim1, Pim3, Trib3, and Serpina 3g.255
The distal end of the EpoR cytoplasmic domain and phosphorylation of Y479 are required for activation of the PI3-kinase/Akt and MAP kinase signaling cascades (Fig. 6.10). Binding of the p85 regulatory subunit of PI3-kinase to EpoR mediates translocation of the kinase from the cytoplasm to phosphorylate lipids at the cell membrane,279 resulting in modulation of survival, metabolism, and translation through the subsequent effects of Akt (a kinase directly downstream of PI3-kinase) on mTOR, GSK3, FOXO, and GATA-1 transcription factors.280,281 The central role of PI3 kinase in signaling downstream of the receptor for Epo is suggested by the ability of a constitutively active Akt partially to rescue erythroid development when expressed in JAK2−/− fetal liver cells.280 With regard to the ras/raf/MAPK cascade, Jak2-initiated phosphorylation of tyrosines at the distal end of EpoR allows a Shc adaptor protein to associate with the receptor, which then recruits GRB2 and SOS, resulting in activation of cell membrane-bound Ras and then Raf activation. This signaling cascade gives rise (via MEK) to phosphorylation of ERK1/2 which subsequently phosphorylates up to 60 substrates, promoting erythroid cell-cycle progression and proliferation.282 In contrast to the two pathways described above, however, knockdown of Ras in erythroid cells has only subtle effects on terminal erythropoiesis.283 Although Epo may act on progenitor cells to promote survival, drive proliferation, and direct erythroid maturation, it is not clear if some intracellular signaling pathways can distinctly activate only one of these events. Epo-dependent activation of the PI3-kinase pathway appears important for both cell survival and proliferation whereas the MAP kinase pathway appears more important in directing proliferation.279
Apart from these three major pathways there are a number of other well-described mediators of responses downstream of Epo/EpoR that likely affect erythropoiesis. These include the inositide-specific phospholipases C (e.g., knockdown of the PLC-γ isoform impedes erythroid development284) and the protein kinase C pathways (e.g., inhibition of the PKCα isoform impairs Epo-induced differentiation, whereas PKCε up-regulation protects erythroid cells from TRAIL-induced apoptosis285,286). In addition, knockdown of the Src family tyrosine kinase Lyn results in attenuated EpoR signaling and decreased erythroid precursor survival.270,287,288 The adaptor SH2-containing proteins CrkL (which is phosphorylated by Lyn and indirectly activates Erk1/2289), Lnk (which attenuates JAK2 signaling290), and Spry1, which appears to down-regulate Erk1/2 and Jak2 activation291 also appear to be important regulators of EpoR signals. Of interest, recent studies indicate that polymeric IgA (pIgA1-oligomers of IgA joined by their J-chains), produced in small amounts by plasma cells, binds to TfR1 present on the erythroblast cell surface. Binding of pIgA1 or Fe-Tf to TfR1 appears to transmit an intracellular signal that results in activation of erythroblast Akt and ERK1/2, stimulating erythroblast proliferation and differentiation.292 This pathway may be important to boost erythroid output during stress erythropoiesis as hypoxia increases pIgA1 levels, or during iron deficiency when Fe-Tf levels are low. Recent studies identify over 160 genes that are significantly affected by Epo/EpoR signaling in murine primary bone marrow derived CFU-E-like progenitors,255 suggesting much remains to be learned.
As is found with other cell signaling cascades there is a need for checks and balances in the form of inhibitory or regulatory factors, to prevent overstimulation of erythroid cells by Epo/EpoR-mediated growth and survival signals. The distal end of EpoR, for example, acts as a negative regulatory domain to which SH2-containing tyrosine protein phosphatases dock (to PY401, 429, 431) to dephosphorylate substrates such as Jak2 and STAT5 and attenuate Epo signaling. For example, a transgenic animal expressing a truncated human receptor Epo developed severe erythrocytosis, mimicking primary familial and congenital polycythemia (PFCP, see Chapter 44) where patients have elevated red cell mass due to mutations in the EpoR gene.293 A number of kindreds with PFCP due to mutations in this distal regulatory region of EpoR have since been reported.294 Identified regulatory phosphatases295 include the SH2-containing tyrosine phosphatases SHP1, SHP2 (PTPN1132), and PTP-1B. Other regulatory factors include the control of EpoR trafficking from the ER by Jak2,296 EpoR internalization upon interaction with Epo,297 proteasomal degradation of EpoR298 and signaling adaptor molecules, and the inducible expression of specific inhibitors such as the suppressor of cytokine signaling protein family members SOCS-1, SOCS-3, and CIS-1 (cytokine inducible SH2-containing protein) by STATs. The SOCS family of proteins down-regulates receptor signaling in a negative feedback manner by (a) competing with STAT5 for binding to EpoR phosphotyrosines and (b) binding and inhibiting the Jak2 kinase activation loop, or (c) ubiquitination and proteosomal targeting of Jak2.299
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


