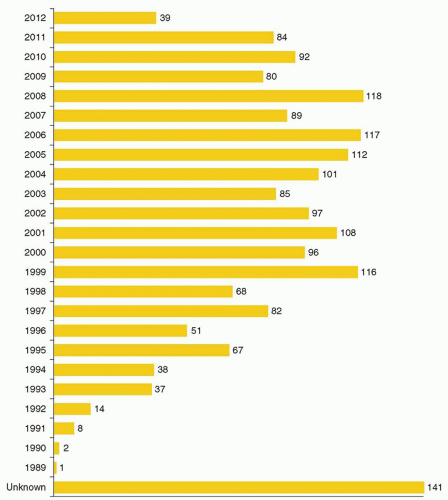
 FIGURE 71.1. Number of gene therapy clinical trials approved worldwide from 1989 through part of 2012. (From The Journal of Gene Medicine, © 2012 John Wiley and Sons Ltd, www.wiley.co.uk/genemed/clinical. Reproduced with permission of John Wiley and Sons, Inc.) |
cells using similar techniques has helped elucidate the in vivo role of these proteins. Significant advances in gene transfer technology have occurred through systematic preclinical testing in large animal models, and clinically relevant gene transfer levels have now been achieved in these models. These results predicted success in humans, at least for disorders for which modest transfer rates of genes not requiring complex regulation could be curative, and unequivocal efficacy has since been realized.22,23 These longanticipated results represented a significant advance for the field, providing not only restoration of immunity in children with severe combined immunodeficiency (SCID), but proof of principle in the context of a human disorder, and a great deal of enthusiasm for returning to the clinic for these and other disorders quickly developed. However, serious adverse events arising as a direct result of insertional mutagenesis have since been reported and efforts to understand these events more fully, develop additional safeguards, and adjust the risk-benefit assessment have now become necessary. Thus, it is particularly important for hematologists to have a general understanding of the field, even if widespread clinical applications may be a decade or more in the future. This chapter reviews the fundamental features of gene transfer technologies and their applications in preclinical and early clinical trials. The pace of the field is rapid, and many details may become obsolete, but the central concepts should remain relevant to any future gene therapy applications.
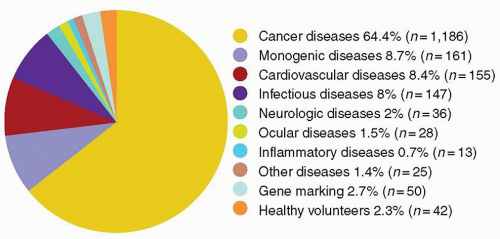 FIGURE 71.2. Indications for gene therapy clinical trials. (From The Journal of Gene Medicine, © 2012 John Wiley and Sons Ltd, www.wiley.co.uk/genemed/clinical. Reproduced with permission of John Wiley and Sons, Inc.) |
A vector may be a simple particle consisting of a fragment of DNA encapsulated within a liposome or conjugated to proteins that facilitate uptake into cells, or may be a more complex viral vector, capitalizing on the ability of viruses to enter cells easily and express genes robustly. Characteristics of the major vector systems are summarized in Fig. 71.4 and Table 71.1, and are detailed in subsequent sections. The successful interaction of a vector with a target cell, leading to an alteration in that cell’s genotype, is termed transduction.
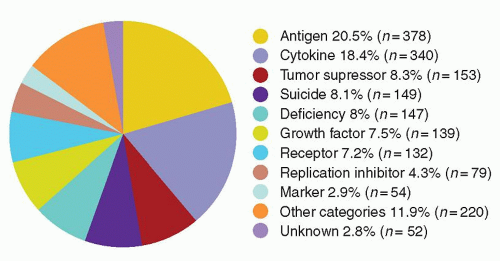 FIGURE 71.3. Gene types transferred in gene therapy clinical trials. (From The Journal of Gene Medicine, © 2012 John Wiley and Sons Ltd, www.wiley.co.uk/genemed/clinical. Reproduced with permission of John Wiley and Sons, Inc.) |
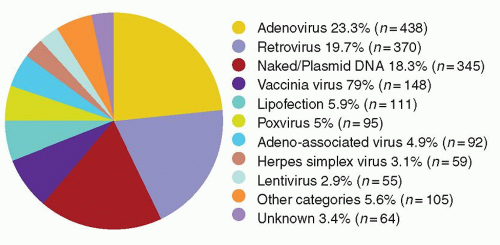 FIGURE 71.4. Vectors used in gene therapy clinical trials. (From The Journal of Gene Medicine, © 2012 John Wiley and Sons Ltd, www.wiley.co.uk/genemed/clinical. Reproduced with permission of John Wiley and Sons, Inc.) |
utility due to the impossibility of injecting enough cells to produce most of the desired effects. One exception is the observation that direct DNA injection into muscle or skin can stimulate a very potent immune response against antigens introduced as plasmid transgenes.44,45 This observation led to preclinical and clinical development of genetic vaccination strategies against bacterial and viral pathogens.46
TABLE 71.1 GENE TRANSFER VECTOR SYSTEMS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
and allow target cell specificity via cell-surface receptors.61,62 For example, adenoviral capsid elements (especially the penton base protein) help disrupt endosomes, releasing DNA more efficiently into the cytoplasm, and inclusion of transferrin or other ligands in DNA-polylysine conjugates allows specific uptake via cell-surface receptors.63, 64, 65, 66 and 67
solution or within cells if cell division allowing nuclear entry and integration does not occur. A number of investigators have tried to increase the likelihood of successful transduction by flowing vector solution continuously over target cells or co-localizing vector and target cells using culture dishes coated with fibronectin fragments.91,92 New methods of concentrating and stabilizing vector preparations are also under development.33,87,93
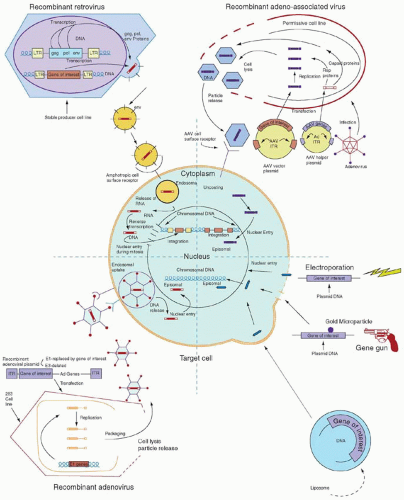 FIGURE 71.5. Schematic representation of transduction of a generic target cell by the four major gene transfer vector systems. The same “gene of interest” is shown being transferred with each vector. AAV, adeno-associated virus; Ad, adenovirus; ITR, inverted terminal repeat; LTR, long terminal repeat. |
efforts. Furthermore, it might be possible to alternate the vector type with subsequent vaccinations to thwart neutralizing antiadenovirus immunity to the vaccine vector. Recently, investigators developing HIV vaccination strategies have reported an adenovirus 41 serotype vector that can express HIV envelope protein.134 The adenovirus 41 serotype virus is an enterotropic pathogen that causes diarrhea in its wildtype form; the use of a gene transfer vector with tropism for a mucosal surface may present advantages for vaccination against HIV, a pathogen that is normally encountered by the host in the mucosa. This vector has not yet been clinically tested.
transient, and production of replication-incompetent vectors has not yet been possible. Very high immunogenicity limits most clinical applications, but may be advantageous for in vivo vaccination with vaccinia-transduced tumor cells.184, 185, 186 and 187
of the common γ-chain expressed in T-cell cytokine receptors (X-SCID),223 and 224,225 JAK3 kinase deficiency,226 deficiency in the ZAP70 protein,227 and recombination-activating gene-2 (RAG-2) deficiency.228
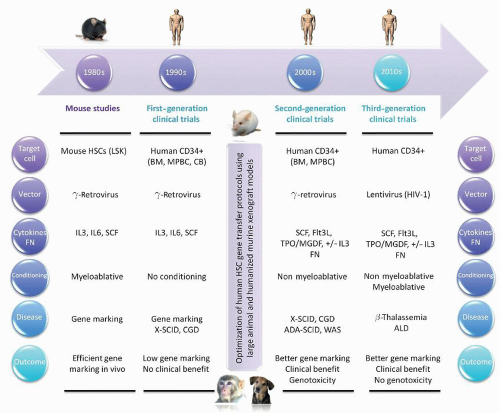 FIGURE 71.6. Timeline for the development of human gene therapy protocols targeting hematopoietic stem cells (HSCs). The main phases in the evolution of HSC gene therapy clinical trials are ordered chronologically. The six principal features defining each phase are outlined. ADA (adenosine deaminase); ALD (adrenoleukodystrophy); BM (bone marrow); CB (cord blood); CGD (chronic granulomatous disease); Flt3L (fms-related tyrosine kinase 3 ligand); FN (fibronectin); HIV (human immunodeficiency virus); IL3 (interleukin 3); IL6 (interleukin 6); LSK (Lin–, Sca1+, Kit+); MGDF (megakaryocyte growth and development factor); MPBC (mobilized peripheral blood cells); SCF (stem cell factor); SCID (severe combined immunodeficiency disease); TPO (thrombopoietin); WAS (Wiskott-Aldrich syndrome). |
retroviral vector and infused in the absence of conditioning.271 Vector sequences were initially detected in circulating myeloid and lymphoid cells at low levels of <0.1% in all three children for >18 months. As expected if corrected lymphocytes have an in vivo advantage, 4 years after the newborns were given infusions of transduced autologous CB CD34+ cells, the frequency of gene-containing T lymphocytes rose to 1% to 10%, whereas the frequencies of other hematopoietic and lymphoid cells containing the gene remained <0.1%. Cessation of polyethylene glycol-conjugated ADA enzyme replacement in one subject led to a decline in immune function, despite the persistence of gene-containing T lymphocytes. Thus, despite the long-term engraftment of transduced HSCs and selective accumulation of gene-containing T lymphocytes, this study indicated that improved gene transfer and expression were needed to attain a therapeutic effect.267
TABLE 71.2 FIRST-, SECOND-, AND THIRD-GENERATION HSC GENE THERAPY CLINICAL TRIALS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


