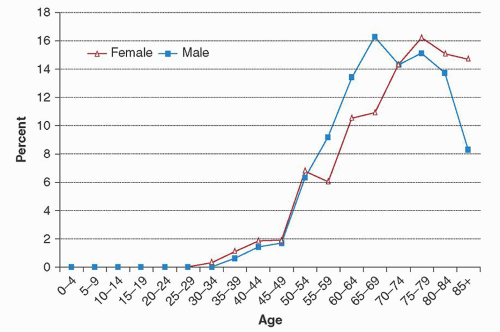
 FIGURE 90.1. Age distribution of 351 males and 265 females diagnosed through the provincial cancer registry and a centralized flow cytometry facility in Manitoba over a 5-year period (1998 to 2003). The median age at diagnosis was 70 years for men and 73 years for women (P = 0.0281). Overall median age was 71.5 years. From Seftel MD, Demers AA, Banerji V, et al. High incidence of chronic lymphocytic leukemia (CLL) diagnosed by immunophenotyping: a population-based Canadian cohort. Leuk Res 2009;33:1463-1468, with permission. |
of HCDR3 with internal epitopes on nearby BCRs.20 As only specific HCDR3 structures may interact with the epitope and induce signaling, this may explain the high incidence of stereotypic HCDR3s. However, no obvious differences in signaling are seen between the BCRs of mutated or unmutated cases or whether the HCDR3 was stereotypic.
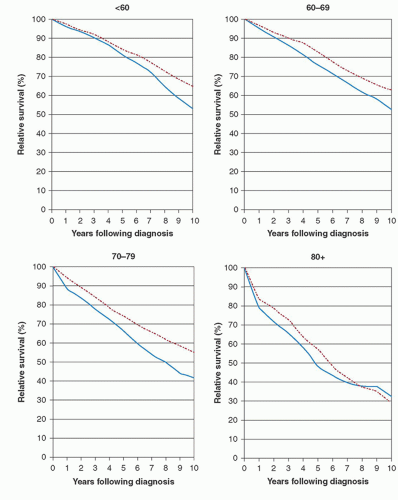 FIGURE 90.2. Ten-year relative survival curves of patients with chronic lymphocytic leukemia (CLL) by major age group. Period estimates for 1980 to 1984 (solid lines ) and 2000 to 2004 (hatched lines). From, Brenner H, Gondos A, Pulte D. Trends in long-term survival of patients with chronic lymphocytic leukemia from the 1980s to the early 21st century. Blood 2008;111:4916-4921, with permission. |
CLL, but a recent study has discounted this hypothesis.37 Patients with familial CLL have also been shown to have elevated plasma levels of B-lymphocyte stimulator (BLγS) and an increased frequency of a C→T polymorphism at -871 in the BLγS promoter.38 Finally, in one CLL family, a single nucleotide polymorphism (SNP) in the promoter site of the death-associated protein kinase 1 (DAPK1) gene was observed in affected family members.39 This polymorphism silenced this gene by binding the transcriptional regulator HOXB7.114. These results suggest that multiple abnormalities may be involved in the development of familial CLL.
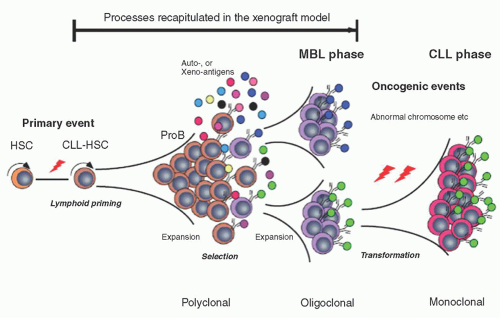 FIGURE 90.3. Schematic presentation of human chronic lymphocytic leukemia (CLL) development based on the xenogeneic transplantation model. CLL human stem cells have accumulated genetic abnormalities that might play a role in amplified B-cell differentiation, and produce a high number of polyclonal B-cells carrying the same genetic aberrations. B-cell clones are selected, and expanded in response to B-cell receptors (BCR) signaling driven presumably by xenoantigens, simulating progression of monoclonal B cell lymphocytosis (MBL). Additional abnormalities such as aberrant karyotypes might play a role in progression from MBL into human CLL. This final step was not recapitulated in the xenograft model. From Kikushige Y, Ishikawa, Miyamato T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011;20:246-259, with permission. |
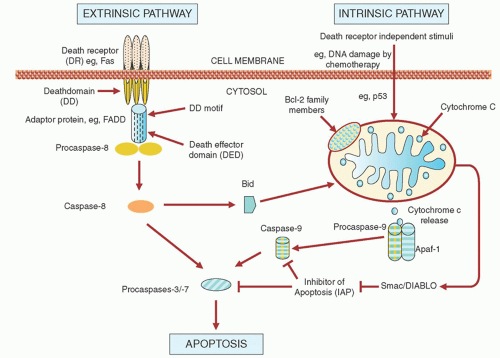 FIGURE 90.4. Apoptotic pathways in chronic lymphocytic leukemia. |
agents may up-regulate receptors for FAS66, 67 or DR4/DR5,68, 69 which could prime the cells to physiologic levels of FAS ligand or TRAIL. However, although fludarabine and chlorambucil increase FAS messenger RNA levels in CLL cells in vitro, they do not affect the levels of FAS protein.60 In contrast, irradiation does increase FAS protein levels, but none of these agents sensitizes CLL cells to FAS ligand.60 Fludarabine and chlorambucil increase the messenger RNA and cell-surface protein expression levels of DR4/DR5 in CLL cells, and this sensitizes the cells to TRAIL.64
immunosuppression and myelosuppression that typify this disease. In addition, the chemokine receptors CXCR4 and CXCR5 on CLL cells regulate CLL cell trafficking to the stromal cells (which produce the receptor ligands, CXCL12 and CXCL13, respectively) in the microenvironment where the CLL cells produce IL-6 and IL-8, which in turn provide signals for cell survival.11, 104 Upon BCR activation, CLL cells secrete cytokines such as CCL3 that attract additional accessory cells such as T-cells to join the CLL and stromal cells.105 In addition to signaling, stromal cells also supply essential nutrients to the CLL cells. For example, CLL cells have limited capacity to transport cystine for glutathione synthesis due to low expression of Xc-transporter.106 In contrast, stromal cells can effectively transport cystine and convert it to cysteine which can then be released into the microenvironment where the CLL cells take up the cysteine for GSH synthesis. CLL cells can also influence stromal cells signaling through microvesicles.107 Microvesicles are released by CLL cells and can modulate the activity of the bone marrow stromal cells (BMSCs). The microvesicle can activate the AKT/mammalian target of rapamycin (mTOR)/p70S6K/hypoxia-inducible factor-1alpha axis in CLL-BMSCs with production of vascular endothelial growth factor (VEGF), a survival factor for CLL cells.107, 108 They can also transfer various messages to target cells that may be critical to disease progression. In the clinic, molecular targeting of the microenvironment through CXCR4 antagonists or BCR kinase inhibitors disrupts the interactions between CLL cells and the microenvironment.109, 110, 111 These chemotherapeutic agents have shown promising pre-clinical activities.
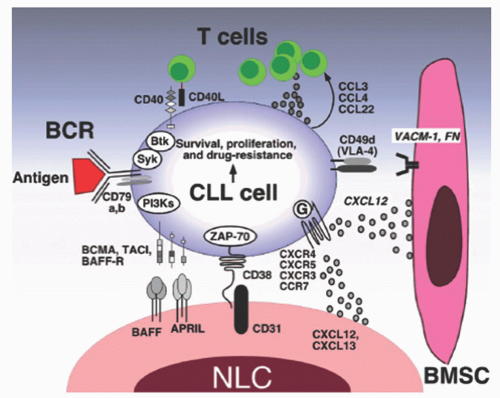 FIGURE 90.5. Molecular interactions in the chronic lymphocytic leukemia (CLL) microenvironment. Molecular interactions between CLL and stromal cells in the BM and/or lymphoid tissue microenvironments that are considered important for CLL-cell survival and proliferation, CLL-cell homing, and tissue retention. Contact between CLL cells and nurselike cells (NLCs) or bone marrow stromal cells (BMSCs) is established and maintained by chemokine receptors and adhesion molecules expressed on CLL cells. NLCs express the chemokines CXCL12 and CXCL13, whereas BMSCs predominantly express CXCL12. The chemokine receptors CXCR3 and CCR7 are additional chemokine receptors on CLL cells that are involved in lymphatic tissue homing. NLCs and BMSCs attract CLL cells via the G-protein-coupled chemokine receptors CXCR4 and CXCR5, which are expressed at high levels on CLL cells. Integrins, particularly VLA-4 integrins (CD49d), expressed on the surface of CLL cells cooperate with chemokine receptors in establishing cell-cell adhesion through respective ligands on the stromal cells (VCAM-1 and fibronectin). NLCs also express the TNF family members BAFF and APRIL, providing survival signals to CLL cells via corresponding receptors (BCMA, TACI, and BAFF-R). CD38 expression allows CLL cells to interact with CD31, the ligand for CD38 that is expressed by stromal and NLCs. Ligation of CD38 activates ZAP-70 and downstream survival pathways. Self- and/or environmental Ags are considered key factors in the activation and expansion of the CLL clone by activation of the BCR and its downstream kinases. Stimulation of the BCR complex (BCR and CD79a,b) induces downstream signaling by recruitment and activation of Syk, Btk, and PI3Ks. Finally, BCR stimulation and co-culture with NLCs also induces CLL cells to secrete chemokines (CCL3, CCL4, and CCL22) for the recruitment of immune cells (T-cells and monocytes) for cognate interactions. CD40L+ (CD154+) T-cells are preferentially found in CLL-proliferation centers, and can interact with CLL cells via CD40. From Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology (Am Soc Hematol Educ Program) 2011;2011:96-103. |
the peripheral blood system. However, NLCs can be found in the spleen and secondary lymphoid tissue.89 There is diverse cross-talk between NLC and CLL cells. NLC express CXCL12, CXCL13, and CD31 plexin B1 BAFF and APRIL and vimentin.88, 112 All these cell-surface markers interact with CLL receptors providing survival and proliferative signals.
co-stimulation of CD40 and FAS on CLL cells can increase apoptosis.154 A novel therapeutic approach has thus been recently explored in which CLL cells are induced to secrete CD40L by a viral transfection strategy.155
activity of the CDKs.186 Normally, when quiescent cells enter the cell cycle, the D cyclins (D1, D2, and D3) in conjunction with CDKs 4 and 6 bring the cell into the S-phase. The D cyclin/CDK4/6 initiates phosphorylation of the retinoblastoma (Rb) protein, which is then further phosphorylated by cyclin E/CDK2. Another function of the D cyclins/CDK4 is the sequestration of the CIP/KIP inhibitors p27Kip1 and p21Cip1, which normally inhibit the activity of cyclin E/CDK2. Although the majority of the peripheral blood CLL cells are quiescent and in G0/G1, the expression of cyclins D1, D2, and D3 are increased in these cells.187, 188, 189 Despite these findings, the Rb protein is not phosphorylated in unstimulated CLL cells190; this may be related to overexpression of p27Kip1, high levels of which are associated with high lymphocyte counts and poor patient survival.191 CDK1 (cdc2) is a serine/threonine kinase involved in G2-M phase transition and gene expression studies have shown that this gene is highly expressed in patients with del 11q22-23 cells.192
TABLE 90.1 INCIDENCE OF GENOMIC ABNORMALITIES AND ASSOCIATED CLINICAL FEATURES IN CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||
deletion (site of the RB1 gene) or translocations with a breakpoint at chromosome 13q14. The 14q abnormalities included translocations from a variety of chromosomes, usually involving chromosome 11, suggesting that these patients had actually mantle cell lymphoma. Less than 5% of patients had an 11q22-q23 deletion. The abnormalities of chromosomes 12 and 13 occurred with equal frequency in cases with a single clonal abnormality and in patients having more than one clonal abnormality.195, 199 In contrast, abnormalities of chromosome 14 occurred primarily in patients having more than one clonal abnormality.195, 199 In general, patients with abnormal karyotyping had a worse prognosis than those with normal cytogenetics, and the outlook was poorest for those with multiple clonal abnormalities.196, 197, 198, 199, 200, 201, 202 In addition, the higher the percentage of cells at metaphase with a clonal chromosomal abnormality, the worse the prognosis. Of patients with a single clonal abnormality, those with trisomy 12 had the worst prognosis, and those with 13q+ had a similar survival as cases with normal karyotyping.199, 200 It is interesting that trisomy 12 was usually seen in those 15% of cases with CLL variants, either CLL/prolymphocytic leukemia (CLL/PLL) or “atypical” CLL, and this may explain the poor prognosis associated with these morphologic variants.202, 203, 204 When all patients were considered, regardless of the number of clones, those with chromosome 14 abnormalities had the worst prognosis.199 As most of the chromosome 14 abnormality group had a t(11;14) (q13;q32), in retrospect, they likely had mantle cell lymphoma and not CLL.3 11q22-q23 deletions have been detected in 13% of patients by karyotyping, and these patients had disease progression and poor survival.205
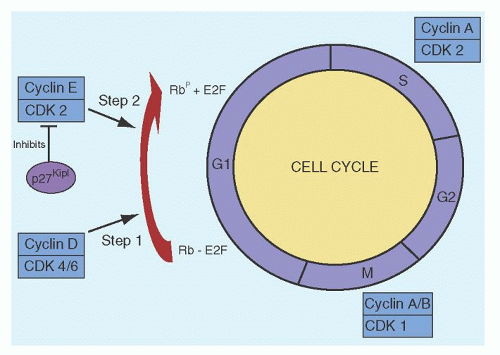 FIGURE 90.6. Control of the cell cycle. Both cyclins D2 and D3 are overexpressed in chronic lymphocytic leukemia cells, but the retinoblastoma (Rb) protein is not phosphorylated, perhaps related to overexpression of p27Kip1. CDK, cyclin-dependent kinase. |
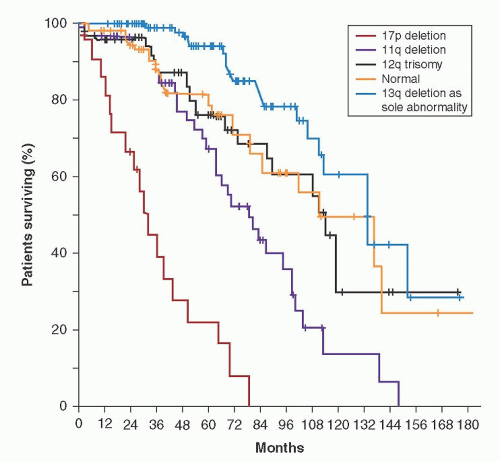 FIGURE 90.7. Survival according to molecular genetic changes in chronic lymphocytic leukemia. From Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910-1916, with permission. |
low-risk FISH and is a useful independent prognostic predictor. Kay et al.219 evaluated genomic complexity using CGH in patients commencing treatment with chemoimmunotherapy. Chromosome deletions were more common than gains and although abnormalties were typically seen in patients with high-risk FISH, complex abnormalities were also seen in patients with 13q deletions. Complex changes were associated with poor response to treatment. Similarly, Ouillette et al.223 have shown that increasing genomic complexity by SNP is an independent prognostic marker, predicting rapid disease progression, poor response to chemotherapy, and short survival. Genomic complexity has also been shown to correlate with resistance of the CLL cells to undergo apoptosis with radiation in vitro.222
TABLE 90.2 IMMUNOPHENOTYPES OF CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) AND OTHER CHRONIC B-CELL DISORDERS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 90.3 SCORING SYSTEM FOR DIAGNOSIS OF CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
in lymph nodes, likely reflecting the higher rate of cell proliferation at that site.115, 265 Although CD38 can be measured easily by flow cytometry there is disagreement about the number of cells that are required to define positivity, with values ranging from 5% to 30%.266, 267, 268 Moreover, the number of CD38 positive cells can vary over time.266, 267, 268
T-cells.133, 134, 135, 136, 137 In addition, CLL cells express both CD40 and the CD40L (CD154), and these cells decrease CD154 expression by normal T-cells with resultant effects on normal B-cell differentiation and isotype switching.309
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


