There probably is no field in medicine that has provided as much hope, or as much disappointment, as the field of tumor immunology. A major relationship between the immune system and the oversight of neoplasms was postulated in the early part of the last century by Paul Ehrlich.1 This theory of immunosurveillance envisioned that, in long-lived animals, inheritable genetic changes in somatic cells must be common, and some proportion of these changes must represent steps toward malignant transformation. It was considered an evolutionary necessity, therefore, that some mechanisms exist for eliminating or inactivating such potentially dangerous mutant cells. This mechanism was thought to be immunologic. The theory of immunosurveillance was restated in the 1950s by Lewis Thomas, then popularized and championed by Sir Macfarlane Burnet.2 Supported by these powerful figures in medicine, the theory of immunosurveillance was so inherently appealing that it often was accepted uncritically, and evidence to the contrary often overlooked.3 For instance, although patients or animals who are immunosuppressed tend to have an increased incidence of tumors, these tumors are disproportionately of lymphoid origin or associated with an oncogenic virus. The development of common epithelial neoplasms (with the exception of certain skin cancers) in these patients occurs with much less impressive frequency.4
The most obvious evolutionary necessity of the immune system was to survey a variety of infections, especially viral infections. Early evidence seemed to indicate that immunity played a significant role in eradicating virally induced tumors.4,5 On the other hand, it appeared to play a less significant, or less effective, role in prevention of tumors induced by physical or chemical carcinogens.6,7
Experimentation in the early part of the 20th century demonstrated that spontaneously arising tumors in outbred animals could occasionally be transplanted from one animal to another of the same species and propagated in that fashion. Attempts to immunize against transplantable tumors soon followed. Animals injected with a small number of tumor cells often were able to eliminate those tumor cells—that is, there appeared to be a threshold number of tumor cells required for tumor propagation. Animals that had eliminated a sublethal inoculum of tumor cells were often able to withstand inoculation with a large number of tumor cells that would have been lethal in a naive animal. Furthermore, preexposure to normal tissue of the donor often rendered the recipient resistant to challenge with a lethal number of tumor cells.8 These experiments brought into question the idea of tumor-specific antigens and ultimately led to the discovery of major histocompatibility complex (MHC) genes and their products.9,10
Modern tumor immunology finds its roots in the classic experiments of Prehn and Main.11 These investigators demonstrated, in genetically identical mice, that previous exposure to a chemically induced sarcoma rendered animals resistant to challenge with the same tumor, but that these animals would accept normal, nonneoplastic tissues transplanted from the tumor donor animal. Similarly, prior exposure to normal tissues from the donor animal did not render the recipient animal resistant to tumor challenge. These experiments revived the notion that tumor-specific (transplantation) antigens did exist. Subsequent experiments demonstrated that protection afforded by prior exposure to tumor cells was tumor specific.12 Thus, the host response to transplanted tumors behaved like an adaptive immune response, demonstrating memory and specificity.
Tumor immunity could be passively conveyed from one animal to another by transfer of lymphoid cells.13 The relevant cells for protection were shown to be T lymphocytes.14 Thus, it should have been clear to workers in the field that the relevant tumor antigens were those that could be recognized by T lymphocytes. However, as this work was beginning there was little understanding of how T lymphocytes recognized antigens or how those antigens were processed and presented to the T lymphocyte by antigen-presenting cells (APCs) or the tumor target cells. Much time and effort were expended in search of membrane structures or tumor cell products that would distinguish the tumor from all others. Particularly after the description of monoclonal antibody technology,15 a fervent search was undertaken to define structures on tumor cells that would be tumor specific and potential targets for therapeutic intervention. Although many cell surface structures were defined, and the contribution to the understanding of biology cannot be overstated, this adventure produced only a single truly specific tumor antigen, the idiotype (Id) of clonally distributed immunoglobulin present on certain lymphomas.
Only recently has convincing evidence for an effect of immunosurveillance been produced.16,17,18 This new evidence relies, in great measure, on the availability of genetically manipulated animal systems. A variety of knockout mice with defects in components of immune activation or effector function develop, at high frequency, spontaneous tumors or tumors after carcinogen exposure. Interferon-γ receptor-deficient mice are more likely to develop methylcholanthrene-induced sarcomas and are more susceptible to spontaneous development of sarcomas and lymphomas after loss of p53 alleles.19,20 One in two aged perforin-deficient mice develops disseminated lymphomas.21 These tumors are rejected by histocompatible wild-type mice through a mechanism dependent on CD8+ T lymphocytes. A high incidence of lymphoma is also seen in aged mice deficient in Fas/Fas ligand interactions.22 Aged mice doubly deficient in signal transducer and activation of transcription (Stat) 1 and recombination activating gene (Rag) 2 develop adenocarcinomas (colon, breast, and lung) with high frequency.23 The frequency and distribution of tumors are increased in the doubly deficient mice over the frequencies and distributions seen in singly deficient mice. Many of these observations have been interpreted as evidence that the primary (both first and predominant) mechanism underlying tumor immunosurveillance is the system of innate immunity.16,17,18
INNATE IMMUNITY AGAINST TUMORS
The innate immune system is a widespread and evolutionarily ancient form of host defense against infection. In recent years, there has been an explosion of information regarding innate immunity, including its role in host defense and its regulation of inflammation and adaptive immunity.24,25,26 The innate immune system is made up of many cells. These include dendritic cells (DCs), macrophages, mast cells, neutrophils, eosinophils, natural killer (NK) cells, natural killer T (NKT) cells, and certain subsets of γδ T cells. Each of these cell types has been implicated in immune responses against tumors.
Phagocytic Cells
Many cells of the innate immune system, including neutrophils, macrophages, and DCs, bear receptors that detect “danger”27 in the form of pathogen-associated molecular patterns (PAMPs). Examples of PAMPs include bacterial lipopolysaccharide, lipoprotein, peptidoglycan, and lipoteichoic acids; bacterial CpG DNA; and viral RNA and DNA. These PAMPs are recognized by a variety of pattern recognition receptors (PRRs) expressed by cells of the innate immune system.26,28 The innate immune system is said to distinguish “infectious nonself” from “noninfectious self.” The PRRs of the innate immune system are encoded in the germline. Unlike genes of the T cell antigen receptor and the immunoglobulins, these genes do not undergo rearrangement. They are fixed and detect critical microbial components. Engagement of PRRs with PAMPs can result in pathogen uptake and/or cellular activation.26,28 One important group of PRRs is the family of evolutionary conserved Toll-like receptors (TLRs), which are critically important for innate immune cell activation. Thirteen TLRs, each with specificity for a different PAMP, have been described in mammals. Other PRR families include the nucleotide-binding domain leucine-rich repeats (NLR) receptors and the caspase recruitment domain helicases.
Neutrophils and macrophages typically exert little antitumor activity, unless these cells are activated by bacteria, their products, or cytokines produced by tumor-specific T cells.29,30 Recent studies have suggested that dying tumor cells or damaged tissues can release damage-associated molecular patterns (DAMPs) that can interact with PRRs and thus serve as danger signals.31 NLR receptors such as NLRP3 (NLR family pyrin domain-containing 3), which form large cytoplasmic signaling complexes called inflammasomes, are thought to play a critical role in detecting DAMPs and might regulate the development of tumors either positively or negatively.32,33 and 34 Macrophages can kill tumor cells using the same mechanisms utilized for killing of microorganisms. These mechanisms include phagocytosis and release of cytotoxic molecules such as reactive oxygen intermediates and nitric oxide.35 Activated macrophages also produce a variety of cytokines. Among these cytokines, tumor necrosis factor (TNF)-α plays a major role in the tumoricidal effects of macrophages in vitro.36
Another important role of phagocytes in tumor immunity is to present tumor antigens to T lymphocytes. DCs (and other APCs such as macrophages) can phagocytose tumor cells and present tumor antigens in the context of MHC molecules and costimulatory signals to T lymphocytes.37
Natural Killer Cells
It has been recognized for a long time that NK cells kill MHC class I-deficient tumor cells in vivo and in vitro.38 However, the identity and characterization of receptors mediating NK activation proved elusive for many years. Activation of NK cells now is understood to be dependent on the balance of activating and inhibitory signals emanating from activating and inhibitory receptors on the NK cell surface.39,40 These receptors fall into two major structural classes, those of the immunoglobulin superfamily (KIRs and LIRs) and those of the C-type lectin-like family (NKG2D, CD94/NKG2A, and Ly49). Most inhibitory receptors (e.g., CD94/NKG2A, KIR, and Ly49) recognize classic or nonclassic MHC molecules. An activating receptor on NK cells, NKG2D (also called KLRK1), has now been shown to recognize a variety of stress-induced MHC class I-like molecules (e.g., Rae-1, H60, and MICA/B). Of note, activated CD8+ T cells and mucosal γδ T cells also express NKG2D. Another activation receptor on NK cells is FcγRIII, which can target NK cells to immunoglobulin G (IgG) antibody-coated tumor cells and induce antibody-dependent cell-mediated cytotoxicity.
NK cells can also discriminate between different allelic variants of MHC molecules.41 This phenomenon was originally identified in the context of the hybrid resistance transplant model in mice, where parental bone marrow grafts were rejected by a subset of host F1 NK cells. When faced with mismatched allogeneic targets, a subset of donor NK cells can sense the missing expression of self-human leukocyte antigen (HLA) class I alleles and mediate alloreactions. These alloreactive NK cells can improve engraftment and control the relapse of acute myeloid leukemia (AML) in mismatched hematopoietic transplants.41,42
Natural Killer T Cells
NKT cells are a subset of T lymphocytes that share receptor structures and functions with the NK cell lineage.43 Prototypical NKT cells, often referred to as invariant (i)NKT cells, express a semi-invariant T cell receptor (TCR), which is specific for glycolipid antigens presented by the MHC class I-like protein CD1d. Although NKT cells express an antigen-specific receptor that is generated by somatic DNA rearrangement, these cells belong to the innate rather than the adaptive arm of the immune system.44 The invariant TCR expressed by NKT cells recognizes a limited set of self- and foreign antigens and, therefore, bears similarity to the PRRs expressed by cells of the innate immune system. Further, NKT cells have a natural, activated phenotype and are unable to generate classic memory responses against their cognate glycolipid antigens.
Mice that are deficient in NKT cells have increased susceptibility to MCA-induced sarcomas, indicating that these cells contribute to natural immunity against tumors.45 NKT cells in mice and humans respond to the marine sponge-derived glycosphingolipid α-galactosylceramide (α-GalCer), which has potent antimetastatic activities in mice.45α-GalCer and related NKT cell antigens are being explored as potential cancer immunotherapies.46
Mucosal γδ T Cells
T cells expressing the γδ TCR are enriched in mucosal tissues such as the mucosa of the gut and skin.47 These mucosal γδ T cells have a highly restricted TCR repertoire, suggesting specificity for a limited set of antigens selectively expressed in their respective epithelial compartments. Like NKT cells, mucosal γδ T cells can be classified as being at the interface between innate and adaptive immunity.44 Epidermal and intestinal γδ T cells express NKG2D and become activated when NKG2D binds to stress-induced MHC class I-related molecules.
γδ T cells play a crucial role in immune surveillance against malignant epidermal cells. The incidence of cutaneous malignancies after treatment with a combination of initiator and promoter carcinogens was substantially increased in mice lacking the TCR δ-chain.48 Activation of γδ T cells required both NKG2D and the γδ TCR, suggesting that engagement of NKG2D with its ligand(s) synergizes with signals received through the autoreactive γδ TCR.
ADAPTIVE IMMUNITY AGAINST TUMORS
The adaptive immune system is composed of B and T cells that express diverse antigen-specific receptors, immunoglobulins, and TCRs, respectively. Diversity of these receptors is generated by somatic DNA rearrangement, in a process referred to as VDJ recombination.49,50 Currently, there is little evidence that adaptive immunity plays a major role in natural immunity against tumors (with the exception of tumors induced by viruses). Although mice lacking T lymphocytes have increased susceptibility to the development of MCA-induced sarcomas, this might be largely due to the lack of iNKT cells and/or γδ T cells.18,51 Nevertheless, it is clear that tumors can induce adaptive immune responses (Fig. 70.1), which can be exploited for the development of cancer immunotherapies.
FIGURE 70.1. Immune responses against tumors. A variety of cells and soluble factors of innate and adaptive immunity can participate in immune responses against tumors. Examples of mechanisms that can suppress immune responses against tumors (i.e., production of TGF-β by tumor cells and induction of Treg cells) are also depicted. Ab, Antibody; FcR, Fc receptor; IFN, interferon; MDSC, myeloid-derived suppressor cell; MHC I, major histocompatibility complex class I; MHC II, major histocompatibility complex class II; NK, natural killer; NKT, natural killer T; PRR, pattern recognition receptor; TAA, tumor-associated antigen; TCR, T cell receptor; TGF, transforming growth factor; TNF, tumor necrosis factor; Treg, regulatory T cell.
Antibodies and B Cells
The role of B cells in regulating tumor immunity remains poorly understood. In some tumor models, B cells appear to be important for priming of T cell responses and tumor resistance, whereas in other models, B cells have an inhibitory effect on the generation of cytotoxic T lymphocyte (CTL) responses and tumor rejection.52 Nevertheless, it is clear that tumor-bearing hosts can produce antibodies against a variety of tumor antigens.53 However, strong humoral responses rarely correlate with tumor resistance. Nevertheless, antibodies can be utilized for immunotherapy of cancer, in particular tumors of hematopoietic origin. Antibodies may kill tumor cells by activating complement and promoting phagocytosis by macrophages. Alternatively, antibody-coated tumor cells may be killed by antibody-dependent cell-mediated cytotoxicity, in which Fc receptor-bearing NK cells, macrophages, or neutrophils mediate the killing. In addition, in certain cases, antibodies may directly interfere with the growth of tumor cells, as illustrated by the beneficial effects of anti-HER-2/neu antibodies against breast cancer, which likely involves downregulation of the HER-2/neu growth factor receptor.54
T Lymphocytes
Classic studies with transplantable tumors have demonstrated a critical role of T lymphocytes in tumor immunity.11 CTLs play a particularly important role in tumor rejection, as these cells can directly lyse malignant cells that display tumor antigens in association with MHC class I molecules.55,56 The importance of CD4+ T cells in tumor immunity is less clear. CD4+ T cells may secrete cytokines that promote the development of CD8+ T cell responses, increase the sensitivity of tumor targets to CTL lysis by inducing MHC class I expression, and activate macrophages. Because of their critical role for the development of tumor immunotherapies, we will briefly describe the mechanisms that lead to the induction of T cell responses to tumors.
Antigen Processing and Presentation
T lymphocytes recognize peptide antigens in the context of MHC molecules. These peptides are derived from two distinct pathways.57 Peptides representing proteins sampled from the extracellular world are generally presented in the context of class II MHC proteins, whereas peptides resulting from intracellular synthesis of proteins are presented in the peptide groove of the class I MHC proteins (Fig. 70.2). The binding cleft of MHC molecules has a β-pleated sheet floor and α-helical sides. An immunogenic peptide must be capable of forming noncovalent attachments to key residues along the cleft and interacting with the T cell antigen receptor with other residues. The MHC contact residues of the peptide tend to be near the amino and carboxy terminal ends of the peptide. The cleft of the class I MHC molecule has closed ends and accommodates only a peptide of proper length, 9 to 11 amino acids. The cleft of the class II MHC molecule, on the other hand, is open-ended and can bind peptides of more diverse lengths, 10 to 30 amino acids, with most being 12 to 19.58
Peptides located in class II MHC molecules are derived from proteins that have been consumed by APCs59 (Fig. 70.2A). The proteins are taken up by phagocytosis, or receptor-mediated endocytosis in clathrin-coated pits, or engulfed by pinocytosis. Once internalized, the antigens are located in membrane-bound vesicles called endosomes. The endosomes then become continuous with lysosomes. The enzymology of the endolysosome has been described in some detail.59,60 There, in an acidic environment, disulfide bonds in proteins are first reduced by the enzyme GILT (gamma interferon-inducible lysosomal thiol reductase), and the resulting products are subsequently cleaved to peptides by proteases, predominantly cathepsins. The endosome fuses with an exocytic vesicle budding from the Golgi apparatus that contains newly made class II MHC molecules associated with invariant chain, which has been shown to play a critical role in the assembly, intracellular transport, and function of MHC class II molecules.61 In addition, a chaperone, HLA-DM, plays a critical role in the loading of peptides onto MHC class II molecules. HLA-DM is a peptide exchange factor that binds with empty and peptide-loaded class II molecules in endosomal and lysosomal compartments.61 In the fused vesicle, peptides are loaded into the class II MHC molecules. Fusion of the endosome with the plasma membrane ultimately displays the class II MHC molecule-peptide complexes on the cell surface.
FIGURE 70.2. Presentation of peptides by major histocompatibility complex (MHC) molecules. A: MHC class II-restricted antigen processing and presentation to CD4 T cells. Exogenous protein antigens are taken up by antigen-presenting cells, disulfide bonds are reduced by the interferon (IFN)-γ-inducible lysosomal thiol reductase (GILT), and the proteins are then degraded in endosomal/lysosomal compartments by cathepsins (Cat). MHC class II α and β are synthesized in the endoplasmic reticulum (ER) and associate there with the MHC class II-associated invariant chain (Ii). The class II/Ii complexes then egress to endosomal compartments, where Ii is degraded by cathepsins, until only its class II-associated invariant chain (CLIP) region remains bound by class II. CLIP is then removed from class II by the human leukocyte antigen (HLA)-DM peptide exchange factor. Finally, class II is loaded with peptide and delivered to the cell surface for presentation to class II-restricted CD4 T cells. B: MHC class I-restricted antigen processing and presentation to CD8 T cells. Cytosolic proteins, derived from endogenously synthesized proteins or from cross-presented antigens (as indicated by the arrow), are degraded by immunoproteasomes that contain the interferon IFN-γ-inducible subunits LMP2 (2), LMP7 (7), and MECL-1 (M). Some of the resulting peptides are further processed in the cytoplasm by peptidases and then transported to the lumen of the endoplasmic reticulum (ER) by the transporter of antigen processing (TAP). Some of the peptides undergo further processing in the ER by ER-associated aminopeptidases (ERAP). Peptide-receptive MHC class I heavy chain (HC)/β2-microglobulin (β2m) heterodimers in the ER associate with a variety of chaperones, including calreticulin (crt), ERp57, and tapasin (tpn). After binding with peptides, class I molecules undergo a conformational change, permitting their egress to the cell surface for presentation to class I-restricted CD8 T cells.
Peptides are prepared for presentation on class I molecules in a different fashion (Fig. 70.2B). These peptides are derived from intracellular protein synthesis.62,63 After protein synthesis, proteins introduced into the cytoplasm become the target of the proteasome, a cytoplasmic organelle whose major function is the degradation of proteins tagged for turnover by the addition of ubiquitin.64,65 During conditions of interferon-γ production such as infection, several proteasome subunits (LMP2, LMP7, and MECL-1) become upregulated and are incorporated into newly assembled proteasomes. Proteasomes that include these IFN-γ-inducible subunits are referred to as immunoproteasomes, whereas those that lack these subunits are called constitutive proteasomes. The IFN-γ-inducible proteasome subunits favor the generation of peptides that have increased affinity for MHC class I molecules.66,67 and 68 In addition to the proteasome, several cytoplasmic peptidases (e.g., TPPII, LAP, TOP) have been implicated in the generation of antigenic peptides, although they can cleave some epitopes as well.65,69 These peptides are then transported into the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) proteins.62 Within the ER, peptides may be further trimmed by ER-resident aminopeptidases (i.e., ERAP1 and ERAP2).65,69,70 Assembly of class I MHC heavy chain molecules with β2-microglobulin requires the presence of peptides. Within the ER, empty MHC class I molecules are associated with a variety of chaperones, including calnexin, calreticulin, ERp57, and tapasin. Tapasin is a transmembrane protein that tethers empty class I molecules in the ER to TAP.71 Emerging evidence suggests that tapasin retains unstable MHC class I molecules within peptide-loading compartments until they bind with high-affinity peptides. The assembled MHC class I-peptide complex transits the Golgi apparatus, proceeds in a vesicle to the cell surface, and is displayed on the cell surface after fusion of the vesicle membrane with the plasma membrane.
There has been a lot of interest in the mechanisms whereby tumor cells initiate CD8+ T cell responses. Few tumors are derived from professional APCs and, therefore, do not effectively prime naive CD8+ T cells. It has now been established that tumor cells can be processed and presented by host APCs, particularly DCs, in a process that is referred to as cross-presentation (Fig. 70.2).72,73 Tumor antigens are then processed inside the APC, and peptides derived from these antigens are displayed on MHC class I molecules for recognition by CD8+ T cells. These APCs also express MHC class II molecules and can prime naive CD4+ T cells, which may be important for the generation of effective CD8+ memory responses. Once tumor antigen-specific CTLs are generated, they can kill tumor cells without the requirement for costimulation. While the precise mechanisms of cross-presentation remain poorly understood,74 the concept of cross-presentation has important applications in the development of tumor vaccines.72,75
T lymphocyte Activation
The goal of antigen processing and presentation is the activation of appropriate T lymphocytes to proliferate, produce cytokines, and promote an immunologic reaction or become cytotoxic cells. Although the interaction of the T cell antigen receptor with antigen-MHC provides specificity of response and initiates the crucial events of activation, the interactions are few and have low affinity.76 The interaction between T lymphocytes and APCs or target cells is initially stabilized by a number of nonspecific receptor-counterreceptor interactions, leading to development of an immunologic synapse with its central supramolecular activation cluster.77,78,79 Chief among these interactions is the coupling of CD2 on the T lymphocyte with lymphocyte function antigen-3 on the APC. Also involved is the interaction of the lymphocyte function antigen-1 molecule with intercellular adhesion molecule-1 and intercellular adhesion molecule-2. Once the cells have been apposed, the specific interaction of the T cell antigen receptor and the antigen-MHC can occur. It now appears that the T cell proceeds toward activation only if certain threshold numbers of TCR-MHC/antigen interactions occur.80
The signal transduction pathways that result in T cell activation have been extensively reviewed,81,82 and 83,84 and we will focus here on the most salient features (Fig. 70.3). Ligation of the TCR with an agonist peptide/MHC complex results in phosphorylation of the cytoplasmic portions of the CD3 and ζ components of the TCR. The cytoplasmic domains of CD3 and ζ contain several conserved peptide sequences called immunoreceptor tyrosine-based activation motifs (ITAMs) that are targets for intracellular protein tyrosine kinases that catalyze the phosphorylation of tyrosine residues in various protein substrates. The tyrosine kinase lck interacts with the cytoplasmic domains of CD4 and CD8 and the tyrosine kinase fyn interacts with the TCR-CD3 complex. Binding of the TCR with peptide/MHC complexes results in receptor clustering, bringing CD4/CD8 and lck in close proximity of the ITAMs within the CD3 and ζ-chains. Lck and fyn subsequently phosphorylate tyrosine residues within the ITAMs, which become docking sites for the ζ-associated protein, ZAP-70, a member of the syk family of protein tyrosine kinases. The bound ZAP-70 then becomes a substrate for lck, and phosphorylation of ZAP-70 results in its activation. Activated ZAP-70 phosphorylates several scaffolding proteins, including LAT (linker of activated T cells) and SLP-76, which, when phosphorylated, serve as docking sites for other proteins that, in turn, activate multiple signaling pathways. One of these signaling pathways involves changes in inositol lipid metabolism. Phospholipase Cγ1 (PLCγ1) becomes tyrosine phosphorylated and activated as it associates with LAT. Activation of PLCγ1 leads to the hydrolysis of a minor membrane lipid, phosphatidylinositol biphosphate (PIP2), to yield inositol triphosphate (IP3) and diacylglycerol (DAG). Each of these products activates downstream events. IP3 induces a rapid increase of free Ca2+ by release from membrane-sequestered Ca2+ stores, whereas DAG and Ca2+ activate protein kinase C (PKC) θ. T cell activation also results in the activation of the ras and rac signaling pathways. Adapter proteins that are activated by phosphorylated LAT and SLP-76 result in the activation of the guanine nucleotide exchange factors SOS (son of sevenless) and vav, which activate the ras and rac signaling pathways, respectively.
FIGURE 70.3. Overview of signal transduction events involved in T lymphocyte activation. Interaction of the T cell receptor (TCR) and coreceptors with major histocompatability complex (MHC)-peptide complexes on antigen-presenting cells (APCs) results in multiple signaling events that lead to the activation of several transcription factors that stimulate expression of numerous genes (e.g., the interleukin-2 [IL-2] gene). Note that the precise interactions between different adaptor proteins that participate in proximal TCR signaling events remain incompletely understood. AP, activated protein; DAG, diacylglycerol; Elk, Ets-like transcription factor; Erk, extracellular signal-regulated kinase; Grb, growth factor receptor-bound protein; IκB, inhibitor of κB; IP3, inositol triphosphate; Itk, interleukin-2-inducible tyrosine kinase; Jnk, Jun N-terminal kinase; Jnkk, Jnk kinase; LAT, linker of activated T cells; Lck, lymphocyte-specific protein tyrosine kinase; Mek, Mapk/Erk kinase; Mekk, Mapk/Erk kinase; NF, nuclear factor; NFAT, nuclear factor of activated T cells; PIP2, phosphatidylinositol biphosphate; PKC, protein kinase; PLC, phospholipase C; RasGRP, Ras guanyl nucleotide-releasing protein; SLP-76, SH2 domain-containing leukocyte protein, 76-kD; Sos, son of sevenless; ZAP-70, ζ-associated protein kinase, 70-kD.
These signaling events ultimately result in the activation of a number of transcription factors, including NFAT, NF-κ B, and AP-1. Cytosolic Ca2+ binds with the Ca2+-dependent protein calmodulin, and Ca2+-calmodulin complexes subsequently activate several enzymes, including the serine/threonine phosphatase calcineurin. Calcineurin then dephosphorylates NFAT, which uncovers a nuclear localization signal that permits NFAT to translocate to the nucleus. Activation of NF-κB is dependent, at least in part, on activated PKCθ. NF-κB is normally found in the cytoplasm in association with a protein called inhibitor of κB (IκB). TCR signals result in phosphorylation of IκB, which is then targeted for degradation by the proteasome. Release of IκB uncovers a nuclear translocation signal in NF-κB that permits its translocation to the nucleus. AP-1 is a transcription factor composed of the proteins Fos and Jun, which are activated by the ras and rac signaling pathways, respectively.
The net result of this extremely complex activation system is the expression of new proteins, the acquisition of functional capacity, or the ability to proliferate. T lymphocyte activation is best understood as a culmination of events leading to IL-2 production.85 The constraints on production of this cytokine are more rigorous than those relevant for production of other gene products (such as IL-2 receptor α-chain and transcription factors). The promoter of the IL-2 gene is made up of a number of binding sites for transcription factors, including two NFAT sites, an NF-κB site, and an AP-1 site.86 The combination of production of IL-2 receptor α-chain and IL-2 provides an adequate stimulus for the T lymphocyte to successfully proliferate, giving rise to the antigen-specific clonal expansion of lymphocytes characteristic of immunologic responses. However, there are extraordinary controls against inappropriate activation of T lymphocytes.87 In addition to a first signal delivered via the T cell antigen receptor complex, full activation of T cells also requires a second signal.88 The best characterized origin of these second signals is the interaction of CD28 on the T lymphocyte surface with its cognate ligands CD80 (B7-1) and CD86 (B7-2) on the APC, the most potent of which is the DC. Failure to receive a second signal can lead the T lymphocyte to undergo anergy or apoptosis. Activation of T cells is also regulated by a variety of negative signals, including inhibitory receptors of the CD28 family such as CTLA-4 (cytotoxic T lymphocyte-associated protein 4), which interacts with CD80 and CD86, and PD-1 (programmed death-1), which interacts with PD-L1 and PD-L2.88
TUMOR-ASSOCIATED ANTIGENS
Tumor antigens are like all other antigens of adaptive immunity. That is, with few exceptions, they are peptides that are presented to T lymphocytes in the cleft of an MHC-encoded protein.55 The nature of peptide antigens responsible for immune responses to tumors has been described in a classic set of experiments.55 In essence, two approaches were used. Neither made assumptions regarding the nature of the antigenic peptides. In the first approach, tumor-derived cloned CTLs were established. Next, a library of tumor complementary DNA or genomic DNA was constructed and used to transfect cells expressing appropriate MHC molecules but lacking the tumor-specific epitope. Transfected cells were tested for their ability to activate the tumor-specific CTLs. The transfected DNA was then recovered and sequenced, thus identifying the gene of origin. In the second approach, MHC molecules were isolated from tumor cells. Subsequently, peptides were eluted from the MHC molecules and fractionated chromatographically. These peptide fractions were then used to load APCs and presented to tumor-specific CTLs. Peptide fractions that stimulated T cell responses were then sequenced using conventional Edman degradation or tandem mass spectrometry. These approaches have revealed some surprising characteristics of tumor-specific antigens. Most tumor-specific antigenic peptides discovered thus far have been derived from proteins not usually expressed in any normal adult tissues89 (with the exception of testis and ovary), such as P1A90,91 and MAGE-1,92,93 or they represent differentiation antigens characteristic of the cellular lineage of the tumor, such as tyrosinase,94,95 and 96 gp100,97,98 and 99 and MART1/Aa100,101 in melanoma.
Early definition of tumor antigens focused on MHC class I-restricted peptides.102 This seemed to be the obvious approach because most tumors express MHC class I structures, but few express MHC class II molecules. Also, the point of immunotherapy was to eliminate tumors—a job for cytolytic cells (i.e., for CD8+ cytotoxic lymphocytes that recognize antigen in the context of class I MHC molecules). Early clinical immunization trials103,104 and 105 demonstrated the feasibility and the potential efficacy of immunotherapy with peptides recognized by CD8+ T cells. However, immune responses were, in general, weak and short-lived. At the same time that the trials were being conducted, there was a growing realization of the importance of CD4+ T cells in the immune response against tumors.102,106,107 Techniques similar to those used to define antigens recognized by CTLs have been used to define antigens for CD4+ T cells. However, these techniques are slow and labor intensive. A genetic targeting expression system has been designed to expedite antigen screening.107 It is likely that incorporation of both MHC class I- and II-restricted epitopes in tumor vaccines will be required to generate potent antitumor responses.108
While this direct approach to tumor antigen recognition has proceeded, other investigators have asked whether certain appealing target proteins could be immunogenic. In particular, molecules involved in the process of malignant transformation provide attractive targets for therapeutic intervention.109 Because loss variants of tumor cells bearing these oncogenic proteins would presumably be nonmalignant,110 an immunologic assault on these proteins might be particularly effective. Evidence has been provided that immune responses to both mutated and overexpressed oncogenic proteins can occur in patients with malignancy or can be elicited in animals. Target oncogenic proteins include mutated Ras,111 HER-2/Neu,112,113 BCR-ABL,114,115 PML-RARα,116 and mutated p53.117
A newer approach to definition of tumor-specific antigen targets for humoral immunity, termed SEREX, has been introduced.118 In the SEREX approach, a complementary DNA library is prepared from a patient’s tumor specimen, packaged into phage vectors, and expressed in bacteria. Recombinant proteins from bacterial clones are transferred to nitrocellulose membranes and identified as relevant antigens by reactivity with IgG antibodies present in the patient’s serum. Early studies defined three classes of antigens: (a) known tumor antigens, such as MAGE-1, MAGE-4a, and tyrosinase; (b) products of known genes, such as restin; and (c) unknown gene products.119,120 Because the cellular and humoral arms of immunity work in concert, it is likely that targets of antibody production will also prove to be targets of cellular immunity. The SEREX method provides a direct approach to the definition of potentially relevant tumor antigens.
While the immune system can protect against the development of tumors, interactions between developing tumors and the host are complex. Tumors often develop means to evade immune responses, and tumors that develop in immunocompetent hosts are often more immunogenic than those that develop in immunodeficient hosts. Finally, it is now also well-recognized that the immune system can play both tumor-suppressing and tumor-promoting roles.
IMMUNE EVASION BY TUMORS
Many tumors have devised ways to evade immune responses.18,56,121 First, tumors may lose expression of the antigens that were recognized by antibodies or CTLs. Second, many tumors downregulate expression of MHC class I molecules, rendering these cells resistant to lysis by CTLs.122,123 Third, tumors may fail to induce effective CTL responses because of the absence of costimulatory molecules and/or resistance to uptake by APCs and cross-presentation.124 Instead of inducing an effective immune response, some tumors may actively promote tolerance induction, by inducing anergy, exhaustion, or deletion of tumor antigen-specific T cells.125,126 This might involve the generation of tumor antigen-specific regulatory T cells (Tregs),127 induction of inhibitory costimulatory molecules such as CTLA-4 and PD-1 on tumor antigen-specific T cells,126 and/or expansion of myeloid-derived suppressor cells, a heterogeneous group of myeloid progenitor cells and immature myeloid cells that can inhibit lymphocyte function.128 Fourth, tumor cells may actively suppress immune responses by secretion of suppressive cytokines such as TGF-β or by expression of the Fas ligand, which may engage with Fas on lymphocytes to induce apoptosis.125 Fifth, the tumor microenvironment, most notably the tumor stroma, may be critical in preventing immunologic destruction of tumor cells by effectively generating an immune privileged site.
Immune Sculpting of Tumors
In 2001, an important study showed that the immune system not only can protect against the development of tumors, but also can influence the quality of tumors—that is, the immune system of the host in which a tumor develops influences the immunogenicity of the tumor.23 These investigators showed that RAG2-deficient mice not only develop MCA-induced tumors at higher frequency, but that a substantial portion of these tumors was spontaneously rejected upon transplantation in syngeneic immunocompetent mice. In sharp contrast, tumors derived from immunocompetent mice usually grew progressively in immunodeficient mice. Thus, these findings demonstrated that the immune system not only protects the host from tumor formation but also sculpts the immunogenicity of the tumors, in a process that is now referred to as cancer immunoediting.17,18,129 Cancer immunoediting has been posited to proceed through three sequential stages: (a) an elimination phase where the immune system recognizes and destroys tumors before they become clinically apparent, (b) an equilibrium phase where tumor cells that escaped the elimination phase are continuously destroyed, with emergence of resistant tumor cell variants due to immune pressure, and (c) an escape phase where tumor cells that have successfully evaded immune responses progressively grow. The cancer immunoediting hypothesis represents an extension or modern version of the immunosurveillance hypothesis.
Tumor-promoting Immune Responses
Discussion of tumor-host interactions would not be complete without at least a mention of the tumor-promoting role of the immune system.130 Many environmental factors, including chronic infections, tobacco smoke, and inhaled pollutants, as well as dietary factors and obesity, are associated with a low-level chronic inflammation and represent risk factors for cancer development. Chronic inflammation can contribute to tumor genesis at all stages, by generating nontoxic stress during the initiation of cancer, inducing cellular proliferation to promote cancer development, and enhancing angiogenesis and tumor invasion to promote cancer progression.130
APPROACHES TO IMMUNOTHERAPY
Immunotherapy is the use of the immune system or its components to target and eradicate tumors. B cell lymphomas are considered to be the most immune responsive of all human cancers.131 They can undergo spontaneous regression,132 and partial responses have been elicited through the use of nonspecific immune activators, such as bacillus Calmette-Guérin and IL-2.133,134 Thus, follicular B cell lymphomas represent excellent candidates for immunotherapy.
Antibody Approaches
The most common form of immunotherapy employed in the treatment of cancer is passive immunotherapy, which involves the administration of manufactured antibodies that target a particular antigen (Table 70.1). Monoclonal antibodies have emerged as a potent and effective molecularly targeted therapy for human cancer, usually in combination with chemotherapy.135,136 Therapeutic mAbs, such as rituximab and alemtuzumab, are examples of a passive approach. Currently, several B lymphocyte antigens, including CD20, CD22, and CD52, have been utilized as targets for immunotherapy. These targets, while found uniformly on lymphoma cells, are also expressed on normal immune system components, such as normal B lymphocytes.134,137,138 Personalized immunotherapy, also referred to as Id vaccine therapy, is a patient- and tumor-specific approach. This modality targets unique protein determinants of the immunoglobulin molecules produced by the malignant B cell clone and does not appear to result in depletion of normal lymphocytes or subsequent impairment of the immune system. This technique stimulates the patient’s immune system to attack the tumor through the use of both the humoral and cellular arms of the immune system. As a result, immunologic memory may be established which could translate into long-term remission. Some of these are being tested in phase II and III trials (Fig. 70.4).139
TABLE 70.1 PERSONALIZED ACTIVE IMMUNOTHERAPY VERSUS PASSIVE IMMUNOTHERAPY
Personalized Active Immunotherapy
Passive Immunotherapy
Tumor-specific
Not tumor-specific
Stimulates host immuneresponse
Does not stimulate host immune response
Induces immunologic memory
Temporary antitumor effect
May produce long-term immunity
Requires retreatment
Induces both the cellular and humoral arms of the immune system
Induces the humoral arm of the immune system only (ADCC, CDC)
FIGURE 70.4. Idiotype as a tumor-specific antigen for B-lymphoma cells. Each B lymphocyte expresses an immunoglobulin molecule on its surface, the idiotype (Id) protein, which is capable of recognizing and binding to a unique antigen. When B lymphocytes undergo malignant transformation, the Id sequences are maintained by the malignant clones and can thus serve as tumor-specific antigen. MHC, major histocompatability complex; TCR, T cell receptor. (Adapted from Vose JM. Personalized immunotherapy for the treatment of non-Hodgkin’s lymphoma: a promising approach. Hematol Oncol 2006;24:47-55.)
Unconjugated Antibodies
Unaltered antibodies have been used since the earliest trials of monoclonal antibody therapy in humans.140,141 and 142 In early trials, success was limited by the absence of suitable tumor cell surface targets, antigenicity of first-generation (murine) mAbs in humans, modulation of the target structure from the tumor cell surface, and poor recruitment of immune effector mechanisms.143,144 However, enthusiasm for this approach was rekindled by the enormous success of genetically engineered, chimeric, or fully humanized versions of mAbs, most notably rituximab for lymphoid malignancies and trastuzumab in solid tumors.144
Rituximab is a chimeric monoclonal antibody with humanized framework and Fc regions. It is directed against the CD20, pan B cell antigen. CD20 is present on pre-B cells and mature B cells, but not on precursor cells or terminally differentiated plasma cells. The function of CD20 remains poorly understood,145 although it has been implicated in B cell activation, regulation of B cell growth, and regulation of transmembrane calcium flux. The antibody fixes human complement and elicits antibody-dependent cellular cytotoxicity (ADCC). Rituximab was approved for use as monotherapy in patients with low-grade or follicular CD20+ non-Hodgkin lymphoma (NHL) in 1997. In the pivotal trial involving 166 patients, reported by McLaughlin et al.,146 patients had relapsed or chemotherapy-resistant disease. Patients received four weekly infusions of rituximab at 375 mg/m2. Rituximab produced a tumor response in one-half of the patients, with a median duration of response of 11.8 months, comparable to the patients’ response duration on the last chemotherapy treatment. Human antichimeric antibody responses were uncommon. The most common adverse experience associated with rituximab was a constellation of acute infusion-related events, including chills, fever, headache, rhinitis, pruritus, vasodilation, asthenia, and angioedema. This syndrome can progress to hypotension, urticaria, bronchospasm, and, rarely, death. The risk is particularly great in patients with high circulating white cell counts.147 Other toxicities were, in general, mild and infrequent.148 Neutropenia and thrombocytopenia were unusual. Circulating B cells were depleted and remained low until recovery at a median of 12 months. Immunoglobulin levels, however, remained normal. No increased incidence of infections was seen. Rituximab was evaluated again in the relapsed, refractory, low-grade, or follicular NHL patient population, using eight weekly infusions rather than four.149 This extended regimen produced a response rate of 57% and a time to progression (TTP) in responding patients of more than 19.4 months. Adverse event reporting was commensurate with the longer treatment period.
Davis et al.150 reported a response rate of 43% and a TTP of 8.1 months in the patients with a significant poor prognostic factor, importantly in patients with low-grade or follicular NHL who had bulky disease, using the standard 4-infusion regimen. These investigators also showed an overall response rate (ORR) of 40% in patients who had progressed after an initial response to rituximab, with an estimated median TTP for responding patients of 17.8 months.151
Rituximab has been used in the first-line treatment of patients with indolent lymphoma, both as monotherapy and in combination with chemotherapy. Hainsworth150 reported the results of rituximab as monotherapy in 39 previously untreated patients. Patients received rituximab × 4 at the usual dose and schedule, with an ORR of 54%. Patients who had responded or who had stable disease were treated with an additional four weekly treatments of rituximab at 6 months intervals to a maximum of 4 treatment cycles. The ORR rose to 72% after the second course of treatment. Progression-free survival (PFS) at 1 year was 77%. The addition of rituximab to cyclophosphamide, hydroxyldaunomycin, oncovin (vincristine), and prednisone (CHOP) chemotherapy produced impressive results. Patients were treated with six infusions of rituximab, one associated with each of six cycles of CHOP. In 40 patients with newly diagnosed (n = 31) or relapsed/refractory (n = 9) low-grade or follicular NHL, rituximab plus CHOP produced an ORR of 95% and complete response (CR) rate of 55%. With a median follow-up of 29 months, median duration of response and median TTP had not been reached. Eight of 18 patients tested for the BCL-2 [t(14;18)] translocation by PCR testing were positive at initiation of therapy. Seven of these 8 patients, after therapy, became negative for the translocation. The authors concluded that the addition of rituximab to CHOP produced benefits in efficacy parameters without significant additional toxicity. Elimination of PCR positivity for the t(14;18) translocation had not been previously reported with CHOP alone.
Rituximab also has been used with success in patients with more aggressive lymphomas. Vose et al.151 reported the results of rituximab plus CHOP chemotherapy (again using 6 infusions of rituximab in association with 6 cycles of CHOP) in 33 previously untreated patients with advanced aggressive B cell NHL. The combination produced an ORR of 94% and a CR rate of 61%. With a median observation time of 26 months, 29 of 31 patients achieving a remission were in continuing remission at the time of the report. Thirteen patients were BCL-2 positive at study entry. Eleven of these 13 patients became BCL-2 negative after treatment, and 10 of the 11 remained BCL-2 negative. The authors concluded that the results were achieved without significant added toxicities above those expected with CHOP. The Groupe d’Etude des Lymphomes de l’Adulte undertook a study to compare the utility of CHOP with that of rituximab plus CHOP in elderly patients with diffuse B cell lymphoma.152 Patients between the ages of 60 and 80 years with untreated, diffuse large B cell lymphoma were eligible for the study. Patients were randomly assigned to receive 8 cycles of CHOP chemotherapy (197 patients) or to receive 8 cycles of CHOP, each given after an infusion of rituximab (202 patients). Rituximab plus CHOP produced a superior rate of remission, 76% versus 63% (P = 0.005). With a median follow-up of 2 years, event-free and overall survivals (OSs) were significantly higher in the rituximab plus CHOP group (P < 0.001 and P = 0.007, respectively). These results were achieved without a significant incremental increase in toxicity. The addition of rituximab to CHOP reduced the risk of treatment failure (risk ratio, 0.54; 95% confidence interval [CI]: 0.44, 0.77) and the risk of death (risk ratio, 0.64; CI: 0.45, 0.89). The addition of immunotherapy to standard chemotherapy had accomplished what 25 years of chemotherapy manipulation had failed to do (i.e., improve on the results of CHOP chemotherapy).153,154 Maintenance rituximab has also been found to prolong event-free survival (EFS) and response duration in follicular lymphoma.155 In subsequent ongoing randomized phase III studies,156,157 rituximab maintenance regimen provided significant PFS and OS at 3-year156 and 4-year157 follow-up assessments in both previously treated156 and untreated157 patients with follicular NHL, compared with no further treatment, though no difference in OS has been reported by other investigators.158
The mechanism by which rituximab produces these impressive results is less clear. A number of possible mechanisms have been considered: initiation of complement-mediated cell lysis, induction of ADCC, and signaling via CD20 leading to programmed cell death and/or sensitization to cytotoxic drugs. Pretreatment lymphoma cells from 29 patients were examined by flow cytometry for expression of complement inhibitors CD46, CD55, and CD59.159 Expression of these cell surface inhibitors of complement activation was not predictive of outcome to rituximab therapy. Considerable evidence suggests that induction of ADCC plays an important role in rituximab’s antilymphoma effects. A rituximab-like antibody for which an IgG4γ framework was substituted for the IgG1 framework of rituximab was incapable of producing B cell depletion in primates.160 Rituximab was relatively ineffective in eliminating Raji B cell implants in FcRγ-/-/nu/nu knockout mice compared to nu/nu mice.161 These mice lack the activating receptor for Fc portions of antibodies, a critical component of the antibody-dependent cell-mediated cytotoxicity mechanism. In patients, response to rituximab has been shown to be associated with homozygosity for the high-affinity allotype of the FcγRIIIa receptor.162 Evidence also exists that rituximab signaling or interference with normal signaling via CD20 may directly induce apoptosis or sensitize cells to the deleterious effects of chemotherapeutic agents.163 A direct, growth inhibitory effect of rituximab, with accompanying apoptosis, on cell lines cultured in the absence of complement was demonstrated.164 Anti-CD20-associated apoptosis has been associated with upregulation of the proapoptotic protein, Bax165 and downregulation of antiapoptotic protein BCL-2 through inactivation of STAT3.166 Downregulation of STAT3 appears to be a result of downregulation of an IL-10 autocrine pathway.167 These changes and/or others may be responsible for increased sensitivity to chemotherapeutic agents.168
In the wake of the success of rituximab, a number of other antilymphoma mAbs have entered the clinic.169 Alemtuzumab is a humanized IgG1κ monoclonal antibody directed against the CD52 cell surface antigen.144 CD52 is expressed on normal and malignant lymphocytes of B- and T cell lineage, as well as NK cells, monocytes, and macrophages. Alemtuzumab is indicated for the treatment of B cell chronic lymphocytic leukemia (CLL) in patients who have been treated with alkylating agents and who have failed fludarabine therapy. The pivotal clinical trial was carried out in 93 patients with fludarabine-refractory CLL .170 Alemtuzumab produced a response rate of 33%. Virtually all of the responders were partial responders; the CR rate was 2%. Median duration of response was 7 months. Median TTP was 4.7 months for the group as a whole; 9.5 months for responders. The most common adverse events were infusion related—most were grade 1 or 2 in severity, including rigors in 90% of patients (grade 3 in 14%), fever in 85% of patients (grade 3 or 4 in 20%), nausea in 53% of patients, and vomiting in 38% of patients. Infusion-associated side effects declined with subsequent infusions. During the study, 28% of patients experienced dyspnea, 17% experienced hypotension, and 3% experienced hypoxia. Overall, 55% of patients developed an infection during the study. Approximately one-half of these infections were considered serious (grade 3 or 4). Septicemia occurred in 15% of patients, and two deaths resulted. Opportunistic infections occurred in 12% of patients. Ten percent of patients died during or within 30 days of treatment—one-third of these were attributed to progressive disease. Twenty-four percent of patients discontinued treatment because of a drug-related side effect. Most patients who discontinued had not responded to therapy. Serious infusion-related events associated with alemtuzumab appear to result from ligation of CD16 on NK cells resulting in what has been termed cytokine storm—release of IL-6, TNF-α, and interferonγ.171 Prolonged immunosuppression after use of alemtuzumab can result in opportunistic infections.172 Treatment schemas now include the routine use of prophylaxis with both antibiotics and antivirals.
To improve on the immunogenicity and efficacy of rituximab, the last few years have seen the development of new generations of anti-CD20 monoclonal antibodies (mAbs) with enhanced antitumor activity resulting from increased complement-dependent cytotoxicity (CDC) and/or ADCC and increased Fc binding affinity for the low-affinity variants of the FcγRIIIa receptor (CD16) on immune effector cells. These second-generation mAbs, such as ofatumumab, veltuzumab, and ocrelizumab, are in clinical development. They are humanized or fully human to reduce immunogenicity, but with an unmodified Fc region. Ofatumumab is a fully human anti-CD20 IgG1 mAb in clinical development for hematologic malignancies and autoimmune diseases. Ofatumumab specifically recognizes an epitope encompassing both the small and large extracellular loops of the CD20 molecule, and is more effective than rituximab at CDC induction and killing target cells. Veltuzumab (IMMU-106, hA20) is a humanized anti-CD20 mAb with complementarity-determining regions similar to rituximab. This antibody has enhanced binding avidities and a stronger effect on CDC compared to rituximab. Ocrelizumab is a humanized mAb with the potential for enhanced efficacy in lymphoid malignancies compared to rituximab because of increased binding affinity for the low-affinity variants of the FcγRIIIa receptor. Third-generation mAbs are also in clinical development. They are also humanized mAbs, but in addition they have an engineered Fc to increase their binding affinity for the FcγRIIIa receptor. Third-generation mAbs also in clinical development include AME-133v, PRO131921, and GA-101 (Table 70.2), with enhanced affinity for the FcγRIIIa receptor and an enhanced ADCC activity compared to rituximab.173
Two other mAbs with potential utility in the treatment of lymphoma are in early clinical development.169,174 Epratuzumab is a humanized IgG1 monoclonal antibody directed against the CD22 antigen. CD22 is a pan-B cell antigen with distribution similar to that of CD20. Epratuzumab has a favorable safety profile in early trials. Approximately 50% of follicular lymphoma patients and 25% of diffuse large-cell lymphoma patients responded in a small phase II trial. Some of the responses have been long-lived. A recent phase II trial testing the safety and efficacy of combining epratuzumab with R-CHOP (ER-CHOP) in untreated DLBCL showed that the addition to standard R-CHOP, E 360 mg/m2 intravenously, administered for 6 cycles in 107 patients, showed similar toxicity to standard R-CHOP. ORR in the 81 eligible patients was 96% (74% CR/CR unconfirmed [Cru]) by computed tomography scan and 88% by positron emission tomography. By intention to treat analysis, at a median follow-up of 43 months, the EFS and OS at 3 years in all 107 patients were 70% and 80%, respectively. Comparison with a cohort of 215 patients who were treated with R-CHOP showed improved EFS in the ER-CHOP patients. ER-CHOP was well tolerated and results appear promising as a combination therapy.175
TABLE 70.2 ANTI-CD20 MONOCLONAL ANTIBODIES (mAbs) APPROVED OR POTENTIALLY USEFUL FOR LYMPHOID MALIGNANCIES
mAb
Company
Antibody characteristics
ADCC
CDC
Direct effects
Comparison with rituximab
Rituximab (Rituxan®, Mabthera®)
Hoffman La Roche
Type I, first-generation mouse/human chimeric IgG1
++
++
+
Rituximab (Rituxan®, Mabthera®)
GlaxoSmithKline plc/Genmab A/S
Type I, first-generation, human IgG1
++
++++
+
Binding to different CD20 epitope; more effective at CDC
Veltuzumab (IMMU-106, hA20)
Immunomedics Inc.
Type I, second-generation, humanized IgG1
++
++
+
Slower off-rate, enhanced binding avidity, a superior CDC
Ocrelizumab
Genentech Inc./Biogen Idec Inc./Chugai Pharmaceutical Co. Ltd/Roche Holding Ag
Type I, second-generation, Humanized fusion IgG1
+++
+/-
+
Binding to different CD20 epitope, enhanced ADCC, reduced CDC, enhanced affinity for FcγRIIIa RIIIa
PRO131921
Genentech, Inc.
Type I, third-generation, humanized fusion IgG1
+++
+++
+
Improved binding to FcγRIIIa, better ADCC, superior antitumor efficacy
AME-133 v (LY2469298)
Lilly
Type I, third-generation, humanized fusion IgG1
+++
++
++
Enhanced affinity for FcγRIIIa, superior ADCC
GA-101 (RO5072759)
Glycart Biotechnology AG, Genentech, F Hoffmann-La Roche Ltd
ADCC, antibody-dependent cellular cytotoxicity; CDC, complement-dependent cytotoxicity; IgG, immunoglobulin G; SMIP, small modular immunopharmaceutical; + indicates low cytotoxicity; ++ indicates intermediate cytotoxicity; +++ indicates high cytotoxicity; ++++ indicates very high cytotoxicity; +/- indicates very low cytotoxicity; – indicates lack of cytotoxicity; ? indicates cytotoxicity unknown.
Reproduced with permission from Robak et al. BioDrugs 2011;25:13-25.
Apolizumab is a humanized IgG1 monoclonal antibody that binds to a variant of the HLA-DR β-chain. The antibody induces complement-mediated lysis, ADCC, and tyrosine phosphorylation signaling events in cell lines in vitro. The antibody binds to approximately 70% of lymphoma specimens. Administration of the antibody to patients results in typical infusion-related side effects. Four of 8 patients with follicular lymphoma responded to apolizumab. A phase I/II dose-escalation study of thrice-weekly apolizumab (1.5, 3.0, 5.0 mg/kg/dose) for 4 weeks in relapsed CLL resulted in significant toxicity and lack of efficacy; thus, further clinical trials of apolizumab were discontinued, as were other trials in lymphoma and solid tumors.176 Milatuzumab (hLL1, IMMU-115; Immunomedics) is a fully humanized mAb specific for CD74, a cell surface-expressed epitope of the HLA class II-associated invariant chain. CD74 plays an important role as an accessory signaling molecule and survival receptor in the maturation and proliferation of B cells by activating the PI3K/Akt and the NF-κB pathways.177 Milatuzumab demonstrated antiproliferative activity in transformed B cell lines, improved survival in preclinical models, and is presently being evaluated for the treatment of several hematologic malignancies.
Inroads are being made in other hematologic cancers. In multiple myeloma, it was recently reported that the cell surface glycoprotein CS1 (CD2 subset 1, CRACC, SLAMF7, CD319) was highly and universally expressed on myeloma cells while having restricted expression in normal tissues. Preclinical studies showed that elotuzumab (formerly known as HuLuc63), a humanized mAb targeting CS1, could induce patient-derived myeloma cell killing within the bone marrow microenvironment using a SCID-hu mouse model and that the CS1 gene and cell surface protein expression persisted on myeloma patient-derived plasma cells collected after bortezomib administration. In vitro bortezomib pretreatment of myeloma targets significantly enhanced elotuzumab-mediated ADCC, both for OPM2 myeloma cells using NK cells or peripheral blood mononuclear cells from healthy donors and for primary myeloma cells using autologous NK effector cells. In an OPM2 myeloma xenograft model, elotuzumab in combination with bortezomib exhibited significantly enhanced in vivo antitumor activity. Elotuzumab is currently in a phase I clinical trial in relapsed/refractory myeloma.178 In AML, one strategy for the development of mAbs targeting human AML stem cells involves first identifying cell surface antigens preferentially expressed on AML LSC (leukemia stem cell) compared with normal hematopoietic stem cells. In recent years, a number of such antigens have been identified, including CD123, CD44, CLL-1, CD96, CD47, CD32, and CD25. Moreover, mAbs targeting CD44, CD123, and CD47 have demonstrated efficacy against AML LSC in xenotransplantation models. Hopefully, these antibodies will ultimately prove to be effective in the treatment of human AML.179
Anti-idiotype Therapy
Much time and energy were expended searching for tumor antigens, particularly after the development of mAb techniques.17 These brute-force immunization, hybridization, and screening procedures yielded little. They defined only a single truly tumor-specific antigen. That was the Id of clonally distributed antibody expressed on the surface of certain B cell lymphomas. Because it represents a unique protein structure within the combining site of the antibody, the Id can serve as an antigen for antibody production. This fact was demonstrated by Sirisinha and Eisen180 in the early 1970s. Furthermore, they demonstrated that an immunologic response to Id could lead to tumor protection.181
Levy and Miller182 have explored the utility of anti-Id strategies in indolent B cell lymphoma over many years. Initially, these investigators raised custom-made anti-Id mAbs183 for passive administration. A first patient with far advanced, chemotherapy-refractory disease received anti-Id mAbs. Gradual reductions in serum Id and tumor volume were noted. The patient then entered a long-term complete remission.184 In an initial series of patients treated with anti-Id therapy, 11 patients were reported.185 In this group, a second near-complete remission, four partial remissions, and five insignificant responses were seen. There was little toxicity associated with administration of the anti-Id antibodies. The most common side effects were chills and fever. Transient shortness of breath, headache, nausea, emesis, diarrhea, and myalgias were occasionally observed. Unusual toxicities included transient leukopenia or thrombocytopenia and transient elevations of hepatic enzymes. Several interesting problems of anti-Id therapy were noted in this early series: the interfering effect of circulating Id, the development of human antimouse antibody, and the emergence of Id-negative tumor cell variants.186,187,188 and 189 An attempt to reduce the incidence of emergence of Id-negative lymphoma variants with a short course of chlorambucil was unsuccessful.190 The cumulative experience suggests that anti-Id therapy can result in a 15% CR rate and a 50% partial response rate. The mechanism of tumor response in these trials remains unclear. However, response in these trials correlated with anti-Id-induced signal transduction events in the lymphoma cells, suggesting that activation of apoptotic pathways may lead to lymphoma cell death after interaction of the anti-Id antibody with the lymphoma surface-bound immunoglobulin receptor.191 This group has turned to active immunization strategies in indolent lymphoma (see section “Immunization Strategies”).
Radioimmunotherapy
The use of immunoglobulin-radionuclide conjugates in cancer treatment represents appropriation of a classic guided-missile strategy. In theory, the antibody homes to its antigenic target and delivers a cytotoxic assault on the cell to which it attaches. Radionuclides offer certain advantages over other cytotoxic agents. They do not have to be internalized. Radioactive particles can deliver their effects over distances of 1 to 5 mm, thus limiting collateral damage to normal tissues while still potentially providing antitumor effects against antigen-negative variants in the vicinity in what has been termed a cross-fire effect. The principles of radiation physics underlying radioimmunotherapy (RIT) have been reviewed by Press and Rasey.192 Radiolabeled antibodies deliver continuous, exponentially decreasing, low-dose-rate radiation. Traditional external beam radiotherapy delivers intermittent, fractionated radiation at higher dose rates. The most commonly used isotopes for RIT have been iodine 131 (I-131) and yttrium 90 (Y-90). These radionuclides kill cells primarily through emission of β particles, resulting in DNA strand breaks. The β particles of Y-90 are more energetic than those of I-131. They affect cells in a radius of approximately 5 mm compared to approximately 1 mm for those of I-131. I-131 also emits γ rays. This allows direct imaging of the distribution of the radioconjugate but raises issues regarding shielding and health care worker and family member safety.
Several recent reviews attest to the evolution of this field.193,194 A number of theoretical and experimentally generated concerns with RIT appear to have been overcome in the successful clinical studies described below. There had been concern that effective delivery of radioimmunoconjugates would be impeded by heterogeneous tumor vasculature, slow diffusion of these large molecules in interstitial spaces, heterogeneous biodistribution in tumor nodules, and high intratumoral pressures.
The two products in clinical use presently, Y-90 ibritumomab tiuxetan (Zevalin) and I-131 tositumomab (Bexxar), are both directed against the anti-CD20 antigen of B lymphocytes, the same structure targeted by rituximab. Both products are based on murine mAbs. Both are administered after infusion of unconjugated anti-CD20 antibodies—rituximab in the case of Zevalin and tositumomab in the case of Bexxar. Both have used nuclear medicine imaging as a preparatory step to administration, but it is no longer required for Y-90 ibritumomab. Simple dosimetry is accomplished for I-131 tositumomab by capturing whole-body gamma counts after infusion of a 5 mCi “dosimetric dose” of the agent. Imaging was carried out in Y-90 ibritumomab tiuxetan-treated patients to assure normal biodistribution. Whole-body dosimetry is carried out in I-131 tositumomab-treated patients to allow calculation of a patient-specific activity (mCi) to deliver a desired total-body dose of radiation (cGy). Both have been studied most extensively in indolent lymphoma and have been approved for treatment of patients with relapsed or refractory follicular, including rituximab refractory, or transformed B (CD20+) NHL.
The approval for ibritumomab tiuxetan rested primarily on two clinical studies. The first was a randomized controlled comparison of the effectiveness of Y-90 ibritumomab tiuxetan to that of rituximab in patients with relapsed or refractory, follicular, or transformed B cell NHL.195 The study involved 143 patients; 73 randomized to Y-90 ibritumomab tiuxetan (single administration), and 70 randomized to rituximab (weekly × 4). The median number of prior therapies was 2. Approximately one-half of the patients failed to respond to or had a TTP of less than 6 months to their last chemotherapy regimen. Y-90 ibritumomab tiuxetan produced a statistically superior response rate (using the response definitions of the International Workshop), 80% versus 56% (P = 0.002). The number of durable responders at 6 months favored Y-90 ibritumomab tiuxetan-treated patients, but the significance of the observation was lost at 9 months and 12 months. Median TTP (estimated by Kaplan-Meier methods) was 11.2 months for patients treated with Y-90 ibritumomab tiuxetan and 10.1 months for patients treated with rituximab (P = 0.173). Grade 3 and 4 nonhematologic adverse events were unusual in both groups. Y-90 ibritumomab tiuxetan produced grade 3 or 4 neutropenia in 57% of patients, grade 3 or 4 thrombocytopenia in 60% of patients, and grade 3 or 4 anemia in 2% of patients. One patient in the Y-90 ibritumomab tiuxetan-treated group developed myelodysplasia. One patient in the rituximab-treated group developed pancreatic carcinoma. The second trial was a phase II experience in 57 patients who had failed to respond to rituximab or had a TTP of ≤6 months.196 These patients had a median of 4 prior therapies, and 74% had bulky tumors (greatest diameter ≥ 5 cm). In this patient population, Y-90 ibritumomab tiuxetan produced a response rate of 74% and CR rate of 15%. The median TTP was estimated at 6.8 months. Grade 4 neutropenia occurred in 35% of patients, grade 4 thrombocytopenia in 9% of patients, and grade 4 anemia in 4% of patients.
The pivotal study for I-131 tositumomab enrolled 60 patients with refractory or transformed low-grade NHL who had been treated with at least two different qualifying chemotherapy regimens.197 Patients must also have failed to achieve an objective response or relapsed within 6 months after completion of their last qualifying chemotherapy (LQC) regimen. Median age was 60 years, and other poor prognostic features included: median of 4 prior therapies, bulky disease, bone marrow involvement, elevated serum lactate dehydrogenase, advanced stage, and transformation from an initial low-grade histology to a higher-grade histology in 38% of the patients. A statistically significant improvement in the primary endpoint was achieved, with a longer duration of response (>30 days) after I-131 tositumomab therapy (n = 26) compared to patients after their LQC (n = 5; P < 0.001). Improvements in secondary efficacy endpoints after I-131 tositumomab compared to those after LQC were also achieved: overall response (47% vs. 12%; P < 0.001), duration of response (11.7 vs. 4.1 months; P < 0.001), and CR (22% vs. 2%; P = 0.002). Fifteen of 60 patients (25%) were classified as long-term responders (patients with a MIRROR Panel-assessed TTP of a year or more). Nine (15%) of the 60 patients remained in CR, with TTP ranging from 41+ to 57+ months.
A second trial examined the incremental benefit of the radioconjugate (I-131 tositumomab) compared to the nonradioactive antibody (tositumomab).198 This study was a randomized, twoarm, open-label, multicenter study that enrolled patients with chemotherapy-relapsed or refractory low-grade or transformed low-grade NHL. Patients were randomized to receive either I-131 tositumomab therapy or unlabeled tositumomab alone. The primary endpoint was a comparison of the rates of CR. Secondary endpoints included ORR, duration of responses, and TTP. A total of 78 patients (18% with transformation) participated in the study. Patients had been previously treated with one to three chemotherapy regimens. One or more therapies must have included an anthracycline, anthracenedione, or alkylating agent. A significant difference was observed for the primary efficacy endpoint. The CR rate was 33% (14 of 42 patients) for the patients treated with I-131 tositumomab compared to 8% (3 of 36) for patients treated with unlabeled tositumomab (P = 0.012). In addition, the ORR was greater after treatment with I-131 tositumomab: 23 of 42 patients (55%) compared to 7 of 36 patients (19%; P = 0.002). Nineteen patients initially treated with the unlabeled antibody crossed over to receive I-131 tositumomab after disease progression. A CR then was observed in 42% (8 of 19 patients) and an ORR in 68% (13 of 19 patients) in the crossover patient population. A total of 20 patients (33%) from the I-131 tositumomab-treated populations, including patients in the crossover arm, were classified as having a long-term response, including ten patients continuing in CR, with TTP ranging from 23+ to 59+ months.
The efficacy of I-131 tositumomab was also evaluated in patients who had progressed after rituximab.199,200 Patients must have had prior treatment with at least four doses of rituximab without an objective response, or to have progressed during or after treatment. Twenty-four patients did not respond to their last treatment with rituximab, and, of the 16 patients who did respond to rituximab, five patients had a duration of response exceeding 6 months. A response occurred in 27 of 40 patients (68%), with a median duration of response of 14.7 months (95% CI; 10.6 months no response). A CR occurred in 12 of 40 patients (30%); the median duration of CR had not been reached (95% CI; 11 months no response). Twenty-four patients had a longer (at least 30 days) duration of response after I-131 tositumomab than after rituximab; 5 patients had a longer duration of response after rituximab than after I-131 tositumomab; 9 patients had equivalent durations of response; and 2 patients were censored (P < 0.001). A total of 14 patients (35%) had a TTP of 12 months or longer. The median PFS was 10.4 months (95% CI, 5.7 to 8.6) for all patients and 24.5 months for confirmed responders (95% CI, 16.8 to not reached [NR]). PFS for 15 confirmed CR patients was NR with an estimated 3 years PFS of 73%. Prior response to rituximab did not significantly affect the confirmed OR rate, duration of response, or median PFS.
Radioiodinated tositumomab and Y-90 ibritumomab tiuxetan have also been used at myeloablative doses, with stem cell rescue in patients with relapsed B cell lymphomas.201,202,203,204 Radioiodinated tositumomab was used initially as a single agent.202 Twenty-five patients were imaged after a tracer dose of radioiodinated tositumomab. Twenty-two of these 25 patients achieved favorable biodistributions (i.e., had tumor doses in excess of doses to normal organs). These 21 patients received therapeutic infusions of radioiodinated tositumomab (345 to 785 mCi) followed by reinfusion of autologous hematopoietic stem cells. All patients achieved bone marrow engraftment (19 with bone marrow stem cells, two with peripheral blood stem cells). However, two patients died before full neutrophil recovery; one of sepsis, one of progressive lymphoma. Nonhematologic toxicities included nausea in most patients, mild mucositis in five patients, and partial alopecia in four patients. One patient experienced reversible cardiomyopathy and interstitial pneumonitis. Eighteen patients responded to this therapy; 16 patients experienced a CR. With a median follow-up of 2 years, 2 years PFS was estimated at 62%, with OS estimated at 93%. Press et al. then combined radioiodinated tositumomab with chemotherapy and autologous stem cell transfusion in a series of patients with relapsed B cell lymphomas.203 Fifty-two patients received the planned therapy. Patients were again given tracer doses of radioiodinated tositumomab and underwent sequential gamma camera imaging. Absorbed doses of radiation to tumor sites and normal organs were determined. Thereafter, patients received a therapeutic infusion of radioiodinated tositumomab calculated to deliver between 20 and 27 Gy to normal organs (e.g., liver, kidneys, and lungs). Patients then received etoposide, 60 mg/kg, and cyclophosphamide, 100 mg/kg, followed by reinfusion of autologous hematopoietic stem cells. The maximal tolerated dose of radioiodinated tositumomab to be combined with chemotherapy was determined to be that dose that delivered 25 Gy to normal organs. Eight patients experienced 13 grade 3 or 4 toxic events. These included 3 patients with acute respiratory distress syndrome, 3 patients with severe mucositis or gastrointestinal toxicity, 1 patient with venoocclusive disease, and 4 patients with fatal infections. At 2 years, the Kaplan-Meier estimates of OS and PFS for all treated patients were 83% and 68%, respectively. These findings were considered superior to results previously observed in patients who had undergone conventional external beam total-body radiation with etoposide/cyclophosphamide preparation for transplantation in the same institution. Thirty-one patients with CD20+ NHL were treated with high-dose Y-90 ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide and were followed by autologous hematopoietic cell transplantation (HCT).204 Treatment also was well tolerated; there were 2 deaths and 5 relapses. At a median follow-up of 22 months, the 2 years estimated OS rates are 92% and 78%, respectively. Retreatment with tositumomab and I-131 tositumomab, has also been found possible in patients with progressive disease after treatment with I-131 tositumomab, who were able to receive subsequent therapy, including cytotoxic chemotherapy and stem cell transplantation.205 In patients with prior response, I-131 tositumomab can produce second responses that can be durable.206
Only gold members can continue reading. Log In or Register to continue
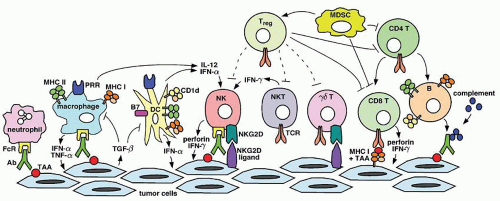
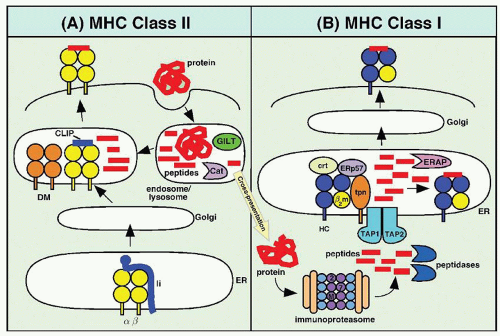
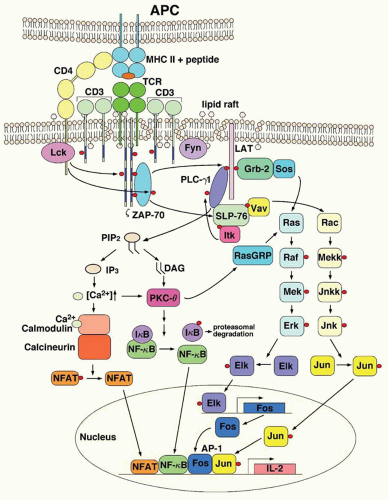
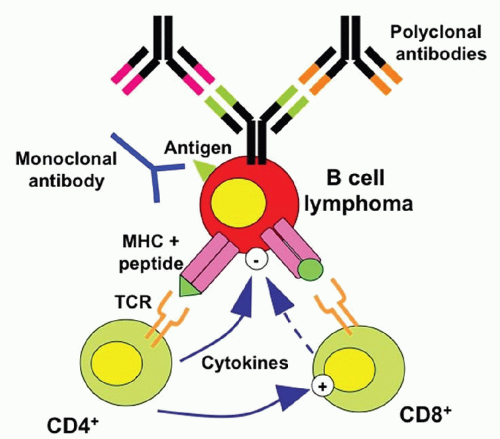
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



