androgen treatment, are also at increased risk of liver tumors. Treatment of malignancies is limited by the low tolerance of FA patients to chemotherapy and radiation. Myeloid malignancies in FA patients have been successfully treated with hematopoietic stem cell transplantation (HSCT), though patient numbers are small and follow-up is limited. The role of pretransplant chemotherapy is unclear.10 For solid tumors, surgical resection is the mainstay of therapy. Regular and frequent surveillance for cancers is particularly important in this population and is discussed further in the section “Supportive Care.” Patients with the FANCD1/BRCA2 subtype manifest an especially high rate of early onset AML and specific solid tumors (brain and Wilms) compared with other FA subtypes.11, 12, 13
TABLE 37.1 PHYSICAL FINDINGS ASSOCIATED WITH FA | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
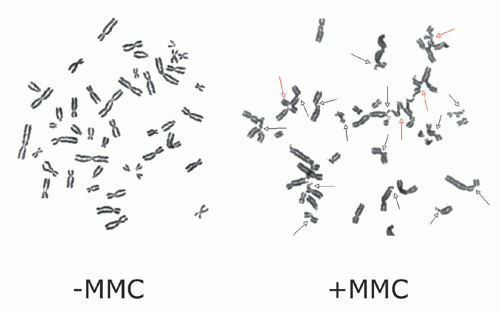 FIGURE 37.1. Chromosomal breakage in FA. Peripheral blood lymphocytes from a FA patient were cultured without (left panel) or with (right panel) mitomycin C (MMC). The black arrows indicate chromosomal breaks. The red arrows indicate radial chromosomal fusions characteristic of FA. (Courtesy of Lisa Moreau, Dana Farber Cancer Institute, Boston, MA.) |
breakage, including the formation of radial figures—a hallmark of this disease. Results are compared to a normal control and a positive control that have been run in parallel. Results are generally reported as aberrations per cell and the number of cells with breaks or radial forms. Increased spontaneous chromosomal breakage may be observed in some FA patients12; nonetheless, the rate of breakage is markedly enhanced by exposure to MMC or DEB regardless of patient phenotype or severity of disease. Chromosomal breakage in response to MMC/DEB can also be assessed in fetal cells obtained for prenatal diagnosis by amniocentesis or chorionic villus sampling.27, 28 If the disease-causing mutation is known for a given family, these assays can be used for prenatal diagnosis or preimplantation genetic diagnosis.29 FA patient cells also exhibit cell cycle abnormalities with G2 phase prolongation and arrest by flow cytometry.30, 31 Studies comparing results of DEB chromosomal breakage with cell cycle analyses of patients referred for FA testing showed close correlation between the results of these two tests.32 Constitutive elevation of serum alpha-fetoprotein (AFP) has been reported in FA patients,33 although variations in AFP levels determined by different methodologies and the lack of specificity has limited the diagnostic utility of this test so far.34 FA heterozygous carriers cannot be reliably detected by testing for chromosomal breakage. Genetic mutation testing is available for the FA genes.35
TABLE 37.2 FA GENES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
with the nuclease FAN1 which can cleave and release the crosslink to allow DNA replication to proceed by translesion synthesis. RAD51 functions in homologous recombination. FANCJ/BRIP1 is a DNA helicase that is important for homologous recombination repair. FANCP/SLX4 interacts with and stimulates the DNA repair endonucleases XPF-ERCC1, MUS81-EME1, and SLX1, and may be important for resolution of homologous recombination intermediates. Elucidation of the Fanconi biochemical pathway in turn led to the identification of interactions between the Fanconi proteins and other known tumor suppressor pathways. These findings provide biochemical support for a role of the Fanconi pathway in tumorigenesis. For example, FANCD2 was subsequently shown to be phosphorylated by ATM kinase at a serine residue at amino acid position 222.46 This phosphorylation is required for the cell cycle S phase checkpoint in response to radiation.46 ATM function is disrupted in the chromosomal instability syndrome ataxia telangiectasia. Thus, FANCD2 links two DNA repair pathways, the Fanconi pathway and the ataxia telangiectasia pathway. Dysfunction of the Fanconi pathway or the ATM pathway is associated with increased cancer susceptibility. The Fanconi biochemical pathway was further implicated in DNA repair by the identification of the gene for FANCD1 as the tumor suppressor gene, BRCA2.43 The FA pathway also interacts with other DNA damage checkpoint proteins including ATR, CHK1, and γ-H2AX.
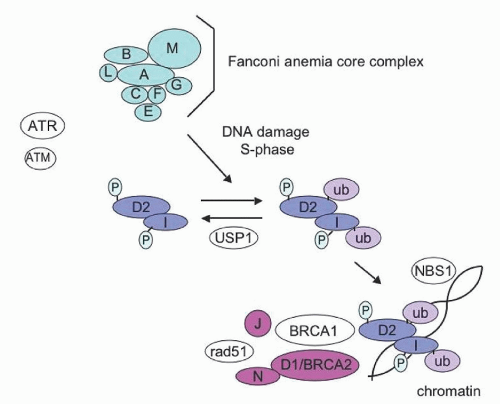 FIGURE 37.2. Schematic diagram of the Fanconi anemia (FA) pathway. FA proteins (A, B, C, D1, D2, E, F, G, I, J, L, M, N) are depicted with shaded circles. Additional DNA repair proteins interacting with the FA pathway are denoted in non-shaded circles. Following activation of the FA core complex by DNA damage or the cell cycle, the D2 and I proteins are monoubiquitinated. Monoubiquitinated D2/I are translocated to chromatin where they colocalize with additional DNA repair proteins to participate in interstrand DNA crosslink repair. Ub, ubiquitin; P, phosphorylation. |
oral cavity and oropharynx is important for patients with FA, DC, and those FA patients previously treated with bone marrow transplantation. Annual endoscopy can be considered in older FA and DC patients. Regular dental exams are important both for maintenance of oral hygiene and for detecting leukoplakia. Patients should be evaluated immediately for symptoms of pain in the mouth or throat, difficulty swallowing, changes in voice, anorexia, or weight loss. Suspicious lesions should be biopsied immediately, since early surgical excision is the mainstay of cancer therapy in FA patients. Annual gynecologic examinations, including Pap smears and HPV (human papilloma virus) exams, are recommended at puberty or after the age of 16. Counseling regarding sexual activity should be provided. Barrier methods of contraception may be particularly pertinent for the FA patients who are already at risk of cervical and vulvar malignancies. Regular breast exams are also recommended, although it is not clear what role mammography should play in cancer screening of these patients. HPV, which may be associated with an increased risk of squamous cell carcinoma of the head and neck, exhibits increased proliferation and epithelial cell expansion secondary to elevated levels of the viral E7 protein when the FA pathway has been disrupted.64 Vaccination against HPV should be offered to all FA patients, males and females, in accordance with current guidelines.
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias During Pregnancy and The Postpartum Period
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias During Pregnancy and The Postpartum Period
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


