Qualitative Disorders of Leukocytes
Ashish Kumar
Keith M. Skubitz
DISORDERS OF PHAGOCYTIC LEUKOCYTES CHARACTERIZED BY MORPHOLOGIC CHANGES
For the most part, the disorders considered here are rare, are usually familial, and often reflect a general metabolic defect, the major manifestation of which may be more serious in tissues than in leukocytes. Nevertheless, the leukocytic morphologic abnormalities may come to the attention of the hematologist, and familiarity with them and the associated diseases may facilitate diagnosis.
Pelger-Huët Anomaly
Pelger-Huët anomaly is a benign anomaly of leukocytes and is inherited as a non-sex-linked, dominant trait. It is characterized by distinctive shapes of the nuclei of leukocytes, a reduced number of nuclear segments (best seen in the neutrophils), and coarseness of the chromatin of the nuclei of neutrophils, lymphocytes, and monocytes. The nuclei appear rod-like, dumbbell-shaped, peanut-shaped, and spectacle-like (“pince-nez”) with smooth, round, or oval individual lobes, contrasted with the irregular lobes seen in normal neutrophils.1 The incidence of this disorder in different studies has ranged from as high as 1 in 1,000 persons2, 3 to 1 in 4,000,4, 5 6,000,6 or even 10,000.7 Originally observed mainly in Holland, Germany, and Switzerland, the anomaly has now been described in other parts of the world, including individuals of Asian8 and African descent and whites. The practical importance of identifying the Pelger-Huët anomaly lies in distinguishing this defect from the shift to the left that occurs in association with infection.
The discovery of this anomaly in rabbits led to breeding experiments and the production of homozygotes.2, 5 These studies demonstrated that in the heterozygote, bilobed, rod-shaped, and spectacle forms predominate, whereas in the homozygote, round nuclei with no evidence of segmentation are predominant. In rabbits, the homozygous form was often lethal, with most animals dying in utero; some survivors suffered skeletal malformations.
TABLE 58.1 DISTRIBUTION OF NUCLEAR LOBES IN NEUTROPHILS OF NORMAL PERSONS AND IN THOSE WITH PELGER-HUËT ANOMALY | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
Two human homozygotes have been reported.2, 9 In them, the cytoplasm of the neutrophils appeared mature, but the nuclei were round or oval in all the neutrophils, in contrast to the fewer than 40% single-lobed neutrophils present in heterozygotes (Table 58.1).2, 6, 9 In the homozygotes, the eosinophils, basophils, and megakaryocytes also were characterized by dense nuclear chromatin and rounded nuclear lobes; these individuals had fewer nuclear lobes than normal subjects.2 Examination of the bone marrow revealed normal morphologic features in myeloid precursors through the myelocyte stage, and electron microscopy revealed persistence of nucleoli in the otherwise mature neutrophils that contained single oval nuclei.9 This finding was interpreted as indicating some retardation of nuclear maturation, because no cytochemical defects were noted in the cytoplasm. This syndrome is now known to be due to mutations in the lamin B receptor (LBR) gene, a gene important in maintaining nuclear structure.10, 11, 12
Pelger-Huët cells appear to be normal functionally,9, 13, 14 are able to phagocytize and kill microorganisms,5 and survive normally in the circulation in both humans15 and dogs.16, 17
The Pelger-Huët heterozygote is recognized by finding (a) 69% to 93% of the neutrophils to be of the bilobed, pince-nez type; (b) few cells with three lobes (usually <10%); and (c) rare or no cells with four lobes (Table 58.1).4, 5, 6 In normal blood smears, no more than 27% of the cells are bilobed, and significant numbers of cells have three or more lobes.5 The presence of similar abnormalities in the blood smear in other family members also is helpful in establishing the diagnosis. In heterozygotes, mature neutrophils with round or oval nuclei of the type that is characteristic of the homozygous state may increase after stress, such as the injection of colchicine18, 3 A shift toward increased numbers of neutrophil lobes was described in a patient with the anomaly who developed pernicious anemia.19
Pseudo- or Acquired Pelger-Huët Anomaly
Cells with morphologic changes, such as those just described, have been observed occasionally in association with myxedema, acute enteritis, agranulocytosis, multiple myeloma, malaria,
leukemoid reactions secondary to metastases to the bone marrow,20 drug sensitivity,21 or chronic lymphocytic leukemia.3 More commonly, pseudo-Pelger-Huët cells (Fig. 58.1)22 are seen in patients with myeloid leukemia of either the acute or chronic type or in those with myeloid metaplasia.23, 24 In these subjects, the pseudo-Pelger-Huët cells tend to appear late in the disease, often after considerable chemotherapy has been administered. In addition, most of the nuclei are of the single oval type characteristic of the homozygous state.23
leukemoid reactions secondary to metastases to the bone marrow,20 drug sensitivity,21 or chronic lymphocytic leukemia.3 More commonly, pseudo-Pelger-Huët cells (Fig. 58.1)22 are seen in patients with myeloid leukemia of either the acute or chronic type or in those with myeloid metaplasia.23, 24 In these subjects, the pseudo-Pelger-Huët cells tend to appear late in the disease, often after considerable chemotherapy has been administered. In addition, most of the nuclei are of the single oval type characteristic of the homozygous state.23
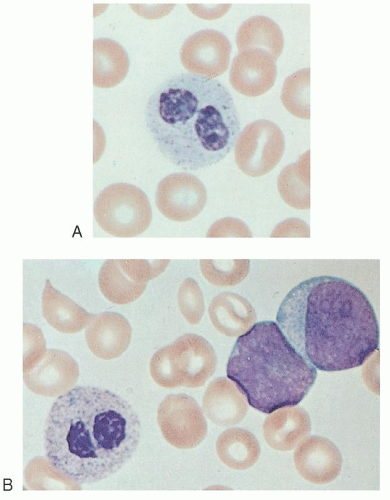 FIGURE 58.1. A: Pseudo-Pelger-Huët cells. B: From a patient with acute myeloblastic leukemia. |
Alder-Reilly Anomaly
Alder-Reilly anomaly, inherited as a recessive trait,25 apparently does not interfere with leukocyte function.26 It is characterized by the presence of larger-than-normal azurophil and basophil granules (Alder-Reilly bodies), which may be easily confused with granulations due to toxic states (Fig. 58.2). These granules stain dark lilac with Wright-Giemsa stains and are seen in patients with various types of bone and cartilage abnormalities.27, 28 They are most common, however, in association with Hurler syndrome, Hunter syndrome, and Maroteaux-Lamy polydystrophic dwarfism.29
Similar inclusions may be seen in blood lymphocytes (Gasser cells) and monocytes.30, 31 The lymphocyte inclusions stain dark red or purple with May-Grünwald-Giemsa stain and metachromatically with toluidine blue; normal azurophilic granules do not stain at all.31, 32 Such lymphocyte granules are found in all types of mucopolysaccharidoses except Morquio syndrome, but they are most common in the Hurler, Hunter, Sanfilippo, and Maroteaux-Lamy syndromes.29, 31, 32 They tend to occur in clusters rather than diffusely throughout the cytoplasm, are surrounded by vacuoles, and are shaped like a dot or comma. In one series of 19 patients, 8% to 50% of the lymphocytes contained the inclusions, and their presence was thought to be of diagnostic significance.32
These inclusions are seen inconsistently in the blood but are more common in the bone marrow. For example, in a series of 18 patients with Hurler form of mucopolysaccharidosis, Alder-Reilly bodies were present in the blood of <10% of them. Careful examination of the bone marrow, however, revealed mucopolysaccharide granules in large mononuclear cells (Buhot cells) in 17 of 18 patients.33, 34, 35
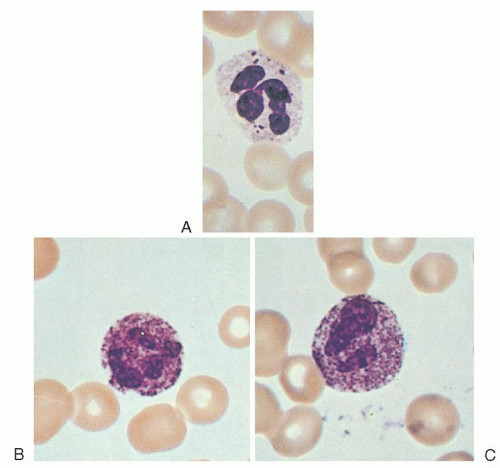 FIGURE 58.2. Neutrophils with Alder-Reilly bodies (A) compared with neutrophils exhibiting toxic granulation (B,C). |
The type of inclusion seen is not diagnostic of a particular type of mucopolysaccharidosis, and the frequency of the inclusions is not correlated with clinical severity.36 The basic defect in this group of diseases lies in the incomplete degradation of the protein-carbohydrate complexes known as mucopolysaccharides, and the different forms of mucopolysaccharidosis involve different enzymatic deficiencies.29 The accumulation of partially degraded mucopolysaccharides within lysosomes has been demonstrated by electron microscopy29; the degradation of the protein core of the mucopolysaccharide appears to proceed normally, but catabolism of the carbohydrate side chains is impaired.
May-Hegglin Anomaly
The May-Hegglin anomaly is a rare, dominantly inherited disorder characterized by large (2 to 5 µm), well-defined, basophilic, and pyroninophilic inclusions in granulocytes (neutrophils, eosinophils, basophils, monocytes), and accompanied by variable thrombocytopenia and giant platelets containing few granules.37 For the most part, affected family members have not been ill, but occasionally abnormal bleeding has occurred.38, 39, 40, 41 Clot retraction time is prolonged, and the reaction to the tourniquet test may be positive. Platelet survival was short (half-life = 3 days, as compared with the normal, which is 6.9 ± 1.5 [1 standard deviation] days).42 Platelet aggregation and retraction were normal, but spreading and serotonin uptake were increased.43 Enzyme and substrate content per platelet were increased, but this value was decreased as related to platelet volume.43 The granulocyte inclusions are similar to Döhle bodies in appearance, but they often are larger, are more round and discrete, and may be present in a large percentage of the cells.40, 41, 44 It is claimed that these inclusions differ from the Döhle bodies of infection in morphologic appearance in that they are present in granulocytes other than neutrophils.44 Like Döhle bodies,40, 44 the inclusions were felt to consist mainly of RNA and may exhibit differing ultrastructure.41, 45, 46, 47 More recently, mutations in MYH9, the gene encoding NMMHC-A,
have been found in this syndrome.48, 49, 50 NMMHC-A is a nonmuscle myosin heavy chain that is part of a family of genes coding for proteins that form part of the actin-myosin force-generating complexes.51 In the bone marrow, clumping of the megakaryocyte cytoplasm has also been reported.39
have been found in this syndrome.48, 49, 50 NMMHC-A is a nonmuscle myosin heavy chain that is part of a family of genes coding for proteins that form part of the actin-myosin force-generating complexes.51 In the bone marrow, clumping of the megakaryocyte cytoplasm has also been reported.39
Mutations in MYH9 are inherited in an autosomal dominant fashion, but depending on the mutation generate a spectrum of clinical findings manifest as several syndromes due to variable expressivity of the clinical features.52, 53, 54, 55, 56, 57, 58 These clinical syndromes include May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome and are characterized by giant platelets, leukocyte inclusions, and variable thrombocytopenia, in addition to problems with hearing, vision, or renal function.52, 53, 54, 55, 56, 57
Chédiak-Steinbrinck-Higashi Anomaly (Congenital Gigantism of Peroxidase Granules)
Chédiak-Steinbrinck-Higashi anomaly, an autosomal recessive disorder, has been reported in humans,59, 60, 61, 62 mice,63 mink,64 cattle,64 cats,65 rats,66 and a killer whale.67 It is characterized by partial ocular and cutaneous albinism, increased susceptibility to pyogenic infections, the presence of large lysosome-like organelles in most granule-containing cells, and a bleeding tendency.68 The abnormal granules are most readily seen in blood and marrow leukocytes, especially granulocytes (Fig. 58.3), and in melanocytes.
In the first families that were reported with this anomaly, the mothers noted that some of their children exhibited pale hair (Fig. 58.4) and photophobia.59, 60 Because these children often had infections and adenopathy and died at an early age, the children born subsequently who were similarly affected were brought to medical attention in the hope of avoiding a fatal outcome.60 It was then that the large, peroxidase-positive granules were noted in their blood and marrow granulocytes.59, 60, 61 Nevertheless, without definitive treatment (bone marrow transplantation, described later) most patients die in infancy or early childhood; only a few survive into early adult life.69, 70, 71
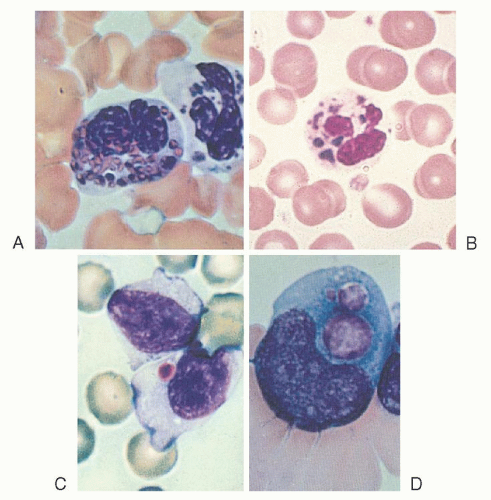 FIGURE 58.3. Inclusion bodies in Chédiak-Steinbrinck-Higashi anomaly. A, B: Neutrophils. C: A lymphocyte. D: A monocytoid cell. |
 FIGURE 58.4. The characteristic silver-gray hair of a child (left) with Chédiak-Steinbrinck-Higashi anomaly contrasted with that of her mother. (Courtesy of Dr. Dorothy Windhorst, National Institutes of Health.) |
During the 30 years after the first description of Chédiak-Higashi syndrome (CHS)72 59 cases were reported.69 The similar disorder in animals is of use in investigations of the pathogenic defect and is of significant economic impact in mink, in that affected animals, inbred for their unusual color, seldom survive more than a year and exhibit increased susceptibility to a slow viral infection that produces hepatitis and a myeloma-like illness (Aleutian mink disease).64
Etiology and Pathogenesis
The manifestations of this disorder appear to result from abnormal intracellular protein transport, resulting in abnormal granule fusion with larger, fewer-than-normal, and perhaps defective granules being formed in most granule-containing cells throughout the body.73, 74, 75, 76, 77 The resulting abnormalities can be found in the hematopoietic tissues, hair, ocular pigment, skin, adrenal glands, pituitary gland, gastrointestinal organs, peripheral nerves, and elsewhere.74
In neutrophils, the anomalously large, peroxidase-positive granules seen by light microscopy59, 60 have been shown by electron microscopy to be abnormal primary (azurophilic) granules, the contents of which remain pleomorphic, the normal granule crystalloid structure not being formed.73, 78 Characterization of the abnormal granules by means of immunofluorescence microscopy demonstrated that they were formed by the progressive aggregation and fusion of azurophilic granules.79, 80
In a subsequent electron microscopic study, investigators demonstrated the presence of two types of giant, peroxidase-positive granules in CHS neutrophils: large, round, or oval granules with homogeneous internal structure similar to normal azurophilic granules; and irregular granules much larger than those of the first type. Because basophils and eosinophils from CHS patients do not contain type 2 granules, and because type 2 granules are less common in promyelocytes, myelocytes, and young neutrophils in the bone marrow than in blood neutrophils, the authors suggested that normal primary (azurophil) granules are formed, but progressive granule fusion occurs during maturation, especially in the blood. In this process, granules and bits of cytoplasm are trapped within the fused granules.81 An abnormal calcium uptake pump in CHS lysosomes may be part of the underlying defect.82, 83 Also noted is an increased tendency to autophagic vacuole formation in Chédiak-Higashi neutrophils, perhaps because of increased permeability and leakage of injurious materials from the massive granules.84 A secretory defect appears to prevent granule release in CHS T cells.85 Abnormal granules have been observed less often in erythroid cells, whereas in megakaryocytes and platelets, the granules appear normal by light microscopy.73 By electron microscopy, however, a decrease in dense bodies was seen,73 and decreased serotonin, adenosine triphosphate, and adenosine diphosphate levels were noted.68
Similar large granules containing a glycolipid have been observed in the Schwann cells of peripheral nerves, in neurons in the central nervous system,84, 86 in renal tubular cells,86 and in the vascular endothelium and fibroblasts.69, 87 In addition, giant pigment granules have been demonstrated in melanosomes86 and in hair strands.69, 86 In the several different tissues affected, the consistent feature is that the histochemical reactions of the large abnormal granules are those usually seen in normal granules of that cell line86; no qualitative alteration in granule enzyme content is noted, but some decrease in granule enzymes and an increase in cytoplasmic enzyme content have been described.88 The effects of the abnormality in different tissues depend on granule function in that tissue. Thus, the large but fewer melanin granules produce pigment dilution, which explains the peculiar hair color, partial albinism, photophobia, and nystagmus.86
The abnormal large granules in neutrophils lead to increased susceptibility to infection. Infection occurs despite an above-normal rate of phagocytosis and a normal postphagocytic metabolic burst (H2O2 production).89 Apparently, the intracellular destruction of some bacteria by Chédiak-Higashi leukocytes is delayed because the postphagocytic delivery of lysosomal enzymes into phagosomes is inefficient and incomplete.90 A similar defect in granule extrusion has been noted in the renal tubule.91
Chédiak-Higashi neutrophils from humans lack cathepsin G and elastase92 and have reduced C3bi receptor expression.93 Because measurements of elastase and cathepsin G were normal in the bone marrow of beige mice, the primary lesion is thought not to be a gene defect but rather is likely a disorder of protein processing, protein synthesis, or granule assembly.92 In addition, a defect in cellular response to chemotactic stimuli both in in vitro and in in vivo skin windows has been demonstrated in humans, mice, and mink,94, 95 and a neutrophil membrane abnormality consisting of spontaneous cap formation has been described.96 Recent studies show that lysosomes function as calcium-regulated secretory compartments and may play an important role in membrane resealing. Lysosomal exocytosis triggered by membrane wounding is defective in human CHS and beige mouse fibroblasts, and this defect may be central to the CHS phenotype.82
Inheritance
In 1972, in families with 127 children, 59 were reported to be affected.69 This statistic and animal studies64 indicate that this disorder is inherited as an autosomal recessive trait. Male and female subjects are affected at a ratio of 0.87:1.00. A high proportion of marriages producing affected children have been consanguineous. Some heterozygotes may be identifiable by the presence of granulation in some of their lymphocytes.97, 98
Clinical Features and Course
The partial albinism (more properly, pigment dilutional defect),86 silvery hair (Fig. 58.4), and photophobia are usually noted early in infancy. The poor resistance to respiratory and cutaneous infection, especially by Staphylococcus and other Gram-positive organisms, soon becomes evident. Four patients studied for more than 1 year experienced 29 episodes of fever and pyogenic infection.71 Increased bleeding with abnormal platelet function has also been recognized.99 Many of the afflicted children develop potentially fatal infections during infancy or early childhood. In others, the disease remains quiescent. Eventually, in more than 85% of patients, it changes to an accelerated phase that is characterized by lymphadenopathy, hepatosplenomegaly, neuropathy, anemia, neutropenia, and, less often, thrombocytopenia.100 During this phase, infiltration of the tissues by mononuclear cells is widespread and the accelerated phase is now considered akin to the disorder hemophagocytic lymphohistiocytosis.101 During the accelerated phase, neurologic manifestations (peripheral neuropathy) may become prominent, and hemorrhage may occur.
Laboratory Findings
The characteristic microscopic findings are the large, often multiple, peroxidase-positive lysosomal granules in the granulocytes of the blood and bone marrow, and the large melanosomes in the hair. Less common are granules in the lymphocytes. Abnormal platelet aggregation can be demonstrated regularly.68 During the early phases of the disease, blood counts yield normal values, but as the disease progresses, anemia, neutropenia, and thrombocytopenia often develop. Immunoglobulin (Ig) and complement levels are normal, as are cellular immune reactions. In the accelerated phase, erythrocyte and granulocyte survival may be shortened.69
Gene Defect
The origin of CHS in mice has been found to be a mutation in the CHS/LYST gene on chromosome 1 in humans, which codes for a lysosomal trafficking regulatory protein.102, 103 The human homologue CHS1 has since been cloned, and mutations of this gene have been found in patients with CHS.76, 77, 102, 104, 105, 106, 107, 108, 109, 110
Management
Some believe that the prophylactic administration of antibiotics may be beneficial.71 The administration of ascorbic acid corrected the defective leukocyte function in vitro and in vivo in several patients,111, 112 and the administration of cholinergic agonists was effective in mice.96, 113 The increased susceptibility to infection has been corrected in mice by the transplantation of normal bone marrow, and the hematopoietic defects can be transmitted to normal mice by transplanting marrow from an animal with CHS.114 In the accelerated phase, splenectomy has been only temporarily helpful69; a few patients have been treated with a combination of vincristine and prednisone. Bone marrow transplantation appears to be a logical treatment, if possible,115, 116, 117 but does not alter the neurologic deterioration.109, 117
Familial Vacuolization of Leukocytes (Jordan Anomaly)
Jordan anomaly is characterized by the presence of vacuoles in the cytoplasm of granulocytes, monocytes, and occasionally lymphocytes and plasma cells. In members of one family, all of the blood neutrophils and more than 70% of the monocytes contained three to ten vacuoles ranging in size from 2 to 5 µm; fewer and smaller vacuoles were seen in eosinophils, basophils, and lymphocytes.118 By means of histochemical and fluorescence microscopic analysis, the vacuoles were shown to contain lipids. These lipids were seen in promyelocytes, myelocytes, metamyelocytes, and occasionally plasma cells in the bone marrow, but they were not present in myeloblasts, erythroblasts, or megakaryocytes.118 This type of vacuolization must be distinguished from that characterized by fat-staining vacuoles occurring in people with serious infections, toxic hepatitis, or diabetic ketoacidosis.119
Other Inclusions in Leukocytes
In an infant with congenital bile duct atresia, amorphous, roundto-oval bodies stained green or gray-green with Romanovsky stains in 3% to 13% of the blood neutrophils and in 1% to 5% of the monocytes.120 Similar inclusions were present in all stages of myeloid cells in the bone marrow but not in lymphocytes or plasma cells. Electron microscopic analysis showed that the inclusions were not enclosed in a phagocytic vesicle.
In the blood monocytes of patients with the Hermansky-Pudlak syndrome,121 a rare familial disorder characterized by albinism, mild bleeding related to platelet dysfunction, accumulation of ceroidlike pigment in marrow macrophages, and deficiency of the tetraspan protein CD63 in platelets,122 lipopigment bodies as well as another type of inclusion were demonstrated.
Abnormal Specific (Secondary) Granule Formation
A syndrome of recurrent staphylococcal skin and sinus infections associated with abnormal chemotaxis, impaired staphylococcal killing, and morphologic abnormalities in the neutrophils was described in a 14-year-old boy.123 No other family members were affected. The patient’s polymorphonuclear neutrophils exhibited bilobed nuclei with unevenly distributed chromatin, drumsticklike nuclear projections, and nearly absent cytoplasmic granules that stained with peroxidase but not with alkaline phosphatase. Electron microscopy revealed primary granules, but specific granules were small and reduced in number. These neutrophils were capable of phagocytosis, generated H2O2, reduced nitroblue tetrazolium (NBT) dye, and killed Candida, but staphylococcal killing was impaired.
Since the original report, at least five cases of specific granule deficiency (SGD) have been identified with both sexes affected, leading to the assumption that the disorder is an autosomal recessive disease.124, 125 SGD neutrophils exhibit decreased chemotaxis and bacterial killing in vitro and decreased migration into skin windows in vivo, and they fail to upregulate CD11/CD18, laminin, or f-met-leu-phe (FMLP) receptors after activation,124 although oxygen radical generation is apparently normal. Myeloperoxidase (MPO)-positive primary granules are present, but the specific secondary granules appeared to be absent in Wright-stained blood smears and were decreased in number and small when viewed with electron microscopy.123 These structures do not stain with alkaline phosphatase. The specific granule constituents lactoferrin and B12 binding protein are reduced or absent.
Lactoferrin deficiency in patients with SGD is tissue specific (i.e., confined to myeloid cells, whereas lactoferrin is secreted normally in nasal secretions) and is secondary to a deficiency of RNA transcripts.126 Because morphologic changes have been described in the primary granule127 and because other granule proteins, such as primary granule defensin92 and “tertiary” granule gelatinase, are also deficient in SGD neutrophils, it was suggested that the basic defect in SGD is one of regulation of production of these proteins.126 Thus, this disorder may be one of incomplete granule synthesis rather than a true SGD.127
C/EBPE is an important regulator of neutrophil secondary granule genes,128 and mutations in the C/EBPE gene have been found in two patients with SGD.129, 130, 131 Neutrophils from Cebpedeficient mice have functional defects very similar to those of patients with SGD,129, 132 and these mice have defects in eosinophil and macrophage function as well.133, 134 Monocyte/macrophage abnormalities have also been noted in an SGD patient.135 One case of SGD has been reported with no mutation in the C/EBPE gene.136
Hereditary Giant Neutrophilia
Neutrophils with a diameter of approximately 17 µm (as compared with a normal diameter of about 13 µm) are rare in blood smears from normal people (1 in 20,000 neutrophils or less). They may be seen with greater frequency in patients who are ill, but even then the number rarely exceeds 0.2% unless a disease involving leukocyte production is present or a reaction to a cytotoxic drug occurs.137 A family with giant neutrophils in healthy members of three generations has been reported.137 Over several years, the propositus had an average of 1.6% giant neutrophils in the blood. The large neutrophils appeared to be nearly double the normal cell volume and contained from six to ten nuclear lobes. Therefore, it was suggested that the cells may have been tetraploid. This anomaly appeared to be transmitted as an autosomal dominant trait.
Hereditary Hypersegmentation of Neutrophil Nuclei
Several families have been described whose members had a hereditary (autosomal dominant) increase in the number of neutrophil nuclear segments (Fig. 58.5).138 The proportion of neutrophils containing five lobes or more exceeded 10% in most heterozygotes and was >14% in several suspected homozygotes, as compared with no more than 10% in normal controls.138 The bone marrow findings suggested a tendency toward nuclear indentation in early myeloid forms (eosinophils and basophils as well as neutrophils).138 The normal size of these neutrophils was thought to provide evidence against tetraploidy, but in one study of five female family members, the mean number of nuclear drumsticks appeared to be increased above normal.139 The chief significance of this anomaly is in its differentiation from other causes of hypersegmentation such as folate or vitamin B12 deficiency.
Hypersegmentation of Eosinophils and Negative Staining for Peroxidase and Phospholipids
Hypersegmentation of eosinophils can be inherited as an autosomal recessive trait, and the abnormality is characterized by a lack of sudanophilia and peroxidase activity in all of the eosinophils, whereas these histochemical reactions remain positive in the neutrophils and monocytes.140, 141 In addition, the number of eosinophilic granules per cell appears to be reduced, and some hypersegmentation of the eosinophil nucleus may occur. No disease accompanies the disorder. This has been most commonly reported in people of Jewish (predominantly Yemenite) extraction, although members of other races have not been studied adequately.
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


