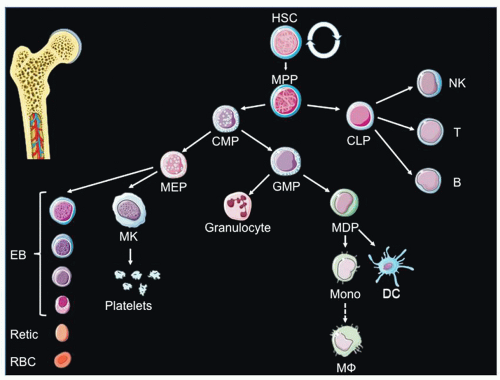
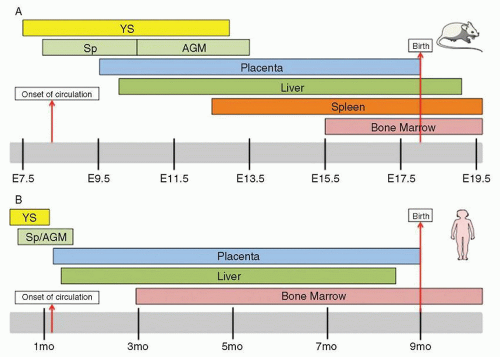
Sites of Hematopoiesis
During prenatal development, the sites of hematopoiesis change several times in the mouse
1,2 and 3 and human
3,4 (
Fig. 5.2). Since better characterized in mice (
Fig. 5.2A) than humans (
Fig. 5.2B), the discussion below will focus on murine developmental hematopoiesis. In humans and other vertebrates, the first hematopoietic cells arise during late gastrulation in the extraembryonic yolk sac (YS) in structures known as blood islands. This initial hematopoiesis is termed
primitive hematopoiesis and serves a supportive role to rapidly produce erythroid cells, platelets, and macrophages prior to the formation of the circulatory system. Primitive hematopoiesis is transient, occurring on embryonic
days 7.25 (E7.25) through 13 (E13) in mice and day 19 through week 8 in humans. Primitive erythrocytes, which are the first embryonic hematopoietic cells, are large nucleated cells morphologically resembling erythrocytes of phylogenetically lower primitive vertebrate groups, such as birds, amphibians, and fish. These primitive erythrocytes have reduced erythropoietin (EPO) requirements during their development compared to definitive erythroid cells
5 that develop later. Also unlike their definitive counterparts, primitive erythrocytes typically circulate as nucleated cells before enucleating, and additionally express
ζ,
βH1, and
εy globin genes.
6,7 These cells and primitive platelets
8 derive from a primitive bipotent megakaryocyte erythroid progenitor found in the yolk sac in mice (E7.25) and humans.
9,10 Along with maternally derived macrophages (M
Φ) that exist, but are declining in numbers, in the yolk sac at E8, two other M
Φ progenitors exist in the yolk sac: one with strictly M
Φ potential
and one with bipotential for M
Φ and erythrocytes.
11 Importantly, since circulation does not commence until E8.25, this indicates in situ M
Φ development in the yolk sac. Thus, primitive hematopoiesis in the yolk sac provides the developing embryo with three crucial hematopoietic cell types prior to contribution from multipotent stem cells deriving from definitive hematopoiesis (see below).
Since the first hematopoietic cells arise in the extraembryonic yolk sac, it was widely believed in the 1970s that the first HSCs developed in the yolk sac. However, experiments in avian chimeras demonstrated for the first time that although the YS had early contribution, the hematopoietic cells present in the stages closer to birth were exclusively derived from the intraembryonic compartment.
12 Similar avian chimeric experiments subsequently demonstrated that the intraembryonic compartment, rather than the YS, was the exclusive source of B and T cells in the adults.
13 Godin and colleagues subsequently demonstrated in mice that the aortic region of E9 embryos, but not YS precursors, were capable of contributing to B cells in irradiated adult recipients.
14 In the same journal issue, Medvinsky, Dzierzak, and colleagues demonstrated that the E10.5 AGM (aorta-gonad-mesonephros) region had substantially higher and earlier onset of CFU-S activity, an early coarse assay for multipotency, compared to YS cells.
15 Soon afterward, Dzierzak’s group demonstrated the ability of E10.5 AGM precursors to provide long-term multilineage reconstitution activity (LTR) in lethally irradiated adult mice.
16 Together, these seminal publications affirmed the intraembryonic contribution to adult mammalian hematopoiesis.
Since the AGM region above was harvested after the establishment of circulation (E8.25), migration of HSCs from a separate undescribed site of origin could not be excluded. To investigate whether HSC development occurred de novo in the AGM, the E8 splanchnopleura (Sp, the future site of the AGM) and the concomitant yolk sac, neither of which have LTR activity, were cultured. While the cultured Sp and YS both produced hematopoietic cells, confirming two independent waves of hematopoietic generation, the YS progenitors were unable to produce lymphoid progeny or have LTR activity.
17,18 Further dissection of the AGM determined that most of the HSC activity is found in hematopoietic intra-aortic clusters found on the ventral wall of the dorsal aorta.
2 HSC activity is also found in the proximal vitelline and umbilical arteries, although these sites have been less characterized. Two reports from the Dzierzak and Mikkola groups established that the placenta represents a previously overlooked major site of hematopoiesis in which HSC emergence parallels that of HSC appearance at E10.5 in the AGM.
19,20 In fact, when LTR HSCs are enumerated, there are 25-fold more LTR HSCs in the placenta than in the AGM.
19 Since the placenta is directly upstream of the fetal liver circulation and since the dramatic expansion of HSC in the FL mirrors that of the placenta, it has been proposed that the placenta is at least a major contributor of LTR HSCs. It has also been proposed that the placenta is a site of de novo HSC emergence independent from the AGM. Indeed, explant and stromal co-culture experiments of mesodermal tissue of the placenta prior to the establishment of circulation demonstrated erythroid and myeloid potential.
21,22 The concept of a de novo generated HSC was bolstered by in vitro culture of E8-9 placenta from
Ncx1-/- animals, which lack a heartbeat and die by E10.5. Without circulatory contribution, the midgestation site had definitive hematopoietic cells with myelo-erythroid and lymphoid potential.
23 Although LTR HSC cannot be isolated from the placenta of
Ncx1-/- animals because of developmental retardation and death by E10.5, these experiments show that definitive hematopoiesis emerges in this organ de novo. Whether the AGM, uterine and vitelline arteries, placenta, or a combination of the above are the genuine origin of HSCs, this LTR activity around E10.5 represents the start of definitive hematopoiesis.
Once definitive hematopoiesis begins, lymphocytes, monocytes, granulocytes, and platelets are formed as well as definitive erythrocytes. At E10, hematopoietic cells (both primitive and definitive) colonize the fetal liver (FL). Dramatic expansion of HSCs occurs at this site (daily doubling in absolute numbers of HSC from E12.5 to E14.5).
24 Eventually, LTR HSCs migrate from the fetal liver to the bone marrow via the circulation, and the bone marrow becomes the primary site of hematopoiesis, with a very small reserve of stem cells remaining in the liver. In the late stages of mammalian fetal development, the bone marrow becomes the main site of hematopoiesis. In humans, the bone marrow is the exclusive site of postnatal hematopoiesis under normal circumstances, whereas in the mouse, the spleen is also a hematopoietic organ throughout life.
Cellular Origin of Hematopoiesis
The cellular intermediates through which mesodermal tissue gives rise to hematopoietic tissue in embryonic development is an area of intense investigation. One candidate cellular ancestor is either (a) a mesoderm-derived bipotent hemangioblast capable of giving rise to either vessels and blood cells or (b) a specialized vascular cell type, called hemogenic endothelium, that serves as a precursor for blood cells. A non-mutually exclusive origin points to HSC derivation from mesenchymal tissue below the endothelial layer. Cytologic analyses of the AGM provide evidence for both endothelial-derived and sub-endothelial-derived HSCs through identification of HIACs and subaortic patches (SAPs), respectively.
2 The strict temporal overlap in the appearance at E10.5 and disappearance at E12.5 of HIACs and SAPs suggests that HSCs derived in SAPs can potentially transendothelially migrate to form HIACs prior to release into the bloodstream.
2 Keller and colleagues definitively showed that a bipotential hemangioblast could be found in the posterior region of the primitive streak in vivo.
25 However, until recently, the existence of a bona fide endothelial intermediate had been under debate. Supportive of an endothelial origin of HSCs is the presence of numerous vessel markers on AGM HSCs, including CD31, VE-Cadherin, and Tie-2.
2 Furthermore, AGM HSCs and endothelial cells in the ventral wall of the E10-E11 dorsal aorta both express Ly6A (Sca-1), c-Kit, CD34, Runx1, SCL, and GATA-2.
1 Fate mapping studies elegantly showed that VE-cadherin expressing endothelium contributes to AGM and adult HSCs, while lineage tracing of subendothelial mesenchyme with Myocardin-Cre animals did not result in labeling of HSCs.
26 Subsequently, novel imaging studies of embryonic stem cell-derived mesodermal cells demonstrated a hemogenic endothelial intermediate in the formation of blood cells.
27,28 Morever, when Runx1, an essential gene in definitive hematopoiesis, was specifically deleted in VE-cadherin-expressing cells (endothelial and hematopoietic cells), but not Vav1-expressing cells (only hematopoietic cells), there was a severe disruption in hematopoietic development that was associated with 65% fetal lethality.
29 Finally, Nancy Speck’s group recently showed that expression of core binding factor beta (CBF
β) in Ly6a-expressing hemogenic endothelium was sufficient for HSC formation.
30 Together, these observations have supported the concept of blood cell development commencing with mesodermal cells that pass through hemangioblastic and hemogenic endothelial intermediates.
Common Critical Genes in Independent Origins of Hematopoiesis
Gene knockout experiments have provided significant insight into the critical regulators of embryonic hematopoiesis. In both primitive and definitive hematopoiesis, Bmp4, Flk1, Tal1
/Scl, Lmo2, Gata2, and Runx1 are all critical for HSC generation.
2 Bmp4 (bone morphogenetic protein 4) is a critical signaling molecule
to specify the dorsal-ventral axis in development. Although the posterior portion of the epiblast in development is fated to give rise to hematopoietic activity, the neurally fated anterior fragment can retain the ability to produce hematopoietic cells by addition of Bmp4.
31 Bmp4 is crucial for hematopoietic development as Bmp4-deficient embryos mostly die around the gastrulation stage, and those that do survive have less yolk sac mesoderm and less lateral plate mesoderm (from which the AGM will develop).
32,33 In definitive hematopoiesis, Bmp-4 is expressed by endothelial cells in the ventral portion of the developing dorsal aorta and the subjacent mesoderm.
2 Using murine ES cells, it was recently shown that Bmp4 is necessary for mesodermal precursor expression of the receptor tyrosine kinase Flk-1 and the bHLH transcription factor Tal-1/SCL.
34The initiation of yolk sac hematopoiesis is dependent on the mesoderm and endoderm layers acting in concert, as soluble factors from endoderm substantially bolster the production of endothelial and hematopoietic cells by murine YS mesoderm explants.
35 One of the candidate soluble factor interactions is VEGF derived from endoderm and its receptor Flk1 on the mesoderm.
36,37 Indeed, Flk-1-deficient embryos do not develop vessels or YS blood islands and die in utero between E8.5 and E9.5.
38 To overcome this early developmental mortality, Shalaby and colleagues performed complementation studies with chimeras of
Flk-1+/- and
Flk1-/- ES cells and demonstrated convincingly that Flk-1 is also required for the generation of definitive endothelial and hematopoietic cells.
39 It was later shown that Flk-1 signaling appears to not be required intrinsically for endothelial and hematopoietic formation, as
Flk1-/- ES cells are able to give rise to endothelial and hematopoietic lineages in vitro
40,41; instead, Flk-1 is likely required for the migration of mesoderm cells from the posterior primitive streak to the yolk sac.
39 In concordance with the importance of the VEGF-Flk-1 signaling axis, VEGF derived from the visceral endoderm (but interestingly, not mesoderm) is sufficient for endothelial and hematopoietic differentiation.
42The transcription factor Tal-1/Scl
43,44 and 45 and the transcriptional regulator Lmo2
46 are both expressed in the yolk sac mesoderm prior to the onset of primitive hematopoiesis and then subsequently expressed in both endothelial and hematopoietic cells. Gene knockout of both Tal-1/Scl
47,48 and Lmo2
49 results in decreased endothelial cells and abrogates YS blood cell production. These genes are also critical for definitive hematopoiesis, as demonstrated by complementation studies with ES cells chimeras.
50,51 and 52 Gata-2-deficient animals have severely impaired primitive hematopoiesis and die at E10.5.
53 Gata2 haploinsufficient embryos have normal yolk sac hematopoiesis,
54 but have a reduction in AGM HSCs, which is consistent with its expression on aortic endothelium
55 and its proposed role in the expansion of hemogenic endothelial progenitors.
55 Runx1 has also been demonstrated to be crucial in definitive hematopoiesis, as Runx1 invalidation abrogates definitive myeloid, lymphoid, and HSC accumulation in the YS, AGM, and fetal liver.
56,57 and 58 Runx1 is thought to be crucial cell autonomously, as complementation studies fail to demonstrate hematopoietic contribution by Runx1-null ES cells.
57 While Runx1 was initially thought to be dispensable in murine primitive erythropoiesis, recent studies have recently shown that the morphology and gene expression of erythrocytes are aberrant in Runx1-deficient animals.
59