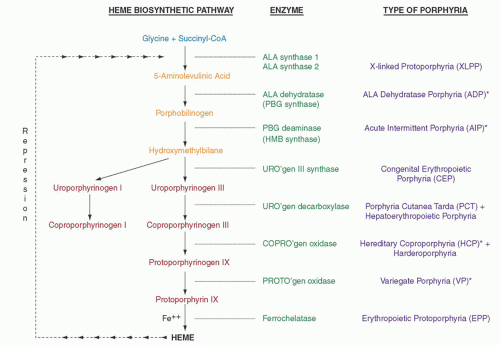
 FIGURE 26.1. The heme biosynthetic pathway, its enzymes, and the eight forms of human porphyria associated with genetic defects of these enzymes. The major compound accumulated and excreted in excess in the seven porphyrias beyond the ALA synthase step is the substrate of the respective deficient enzyme; genetic defects in the erythroid ALA synthase (ALAS2) causing increased enzyme activity lead to increased flux of intermediates through the pathway and accumulated protoporphyrins. In the acute or inducible porphyrias (*), increased hepatic ALA production results from release of the normal negative feedback on ALA synthase exerted by heme,1 indicated by the trail of arrows. ALA, 5-aminolevulinic acid; Fe++, ferrous iron. |
TABLE 26.1 CLASSIFICATION OF THE PORPHYRIAS | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
TABLE 26.2 GENETIC AND METABOLIC FEATURES OF THE PORPHYRIAS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
patients with the disease), the activity of the synthase is intermediate (approximately 50%) between that found in affected individuals and that in normal subjects.8 The UROS gene resides on chromosome 10 (10q25.2-q26.3)9 and has alternative promoters that generate identical housekeeping and erythroid-specific transcripts.7 Forty-six distinct mutant alleles have been identified and molecular defects include point mutations, deletions, insertions, splicing defects, intron branch chain mutations, and erythroid-specific promoter mutations.5, 7, 10 Approximately one third are homoallelic and about 20% of alleles remain undefined. The most common mutation (Cys73Arg) has occurred in one third of alleles (and in nearly one half of homoallelic cases) and correlates with the most severe phenotype of nonimmune hydrops fetalis, transfusion-dependent anemia from birth, or both, particularly in homoallelic cases.5 In most instances, mild forms of the disease have been heteroallelic or only one mutant allele could be identified. Residual enzyme activity of the mutant proteins expressed in prokaryotic systems, or gene promoter-reporter activities in cases of promoter mutations, have provided more precise genotype/phenotype correlations.5, 7, 11 However, in two instances marked divergence of phenotypic expression among siblings suggested undefined modifying factors.12, 13 In another family it was demonstrated that the clinical phenotype is markedly affected by co-inheritance of an activating (gain-of-function) mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene.14
photosensitivity.26, 27, 28 However, this operation is not always beneficial32 and is not always warranted by the degree of anemia. Suppression of erythropoiesis by transfusion decreases porphyrin excretion.5, 33 A young boy with CEP was successfully treated with chronic hypertransfusion;34 transfusion-induced iron overload was avoided by means of concomitant iron chelation therapy. After puberty, his relapse on this regimen was controlled with the addition of hydroxyurea.35
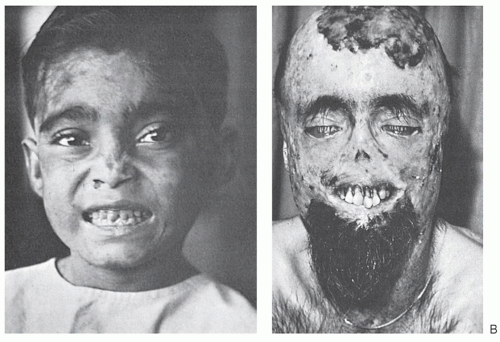 FIGURE 26.2. Congenital erythropoietic porphyria. A: In an Indian boy. Note facial hypertrichosis, scarring, and discoloration of the teeth. B: In a 50-year-old Caucasian man. Note the severe photomutilation. (Courtesy of Dr. Neville Pimstone, University of California, Davis, CA.) |
hepatocyte generated by the common environmental risk factors accompanying clinical expression of the disorder, as well as other traits such as the allele variants of CYP1A2 and G51M1A,105 probably play an important role.94
III.20, 89 Photosensitive cutaneous symptoms rarely occur when the daily urinary excretion of uroporphyrin is <1,000 µg. Urinary coproporphyrin rarely exceeds 600 µg daily.89 An unusual and distinctive tetracarboxylic porphyrin, isocoproporphyrin, is excreted in feces,129 and a slight increase may be noted in total fecal porphyrin excretion.20 Plasma porphyrin level is typically increased, with a fluorescence peak at neutral pH near 619 nm. Homozygotes for the defect have mild anemia; it was severe in three cases.75 The erythrocyte protoporphyrin was found to be moderately increased when measured and is primarily zinc protoporphyrin.75, 78
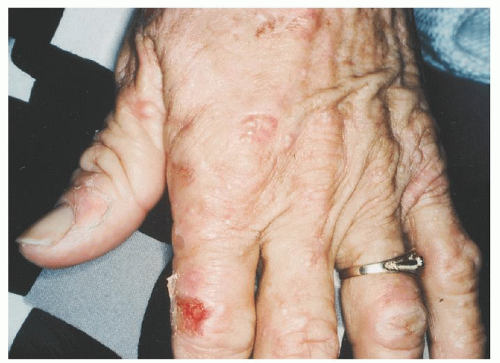 FIGURE 26.3. Porphyria cutanea tarda in a 60-year-old man. Note denuded skin areas over the fingers, an erosion, blisters, and milia. |
of this class inhibit hepatic UROD activity.158 As in the case of hexachlorobenzene-induced porphyria, iron magnifies the inhibition of UROD, and iron depletion minimizes the inhibitory effect of these agents.159 The parallel between the permissive effects of iron in these toxic porphyria models and the role of iron in the pathogenesis of both familial and sporadic PCT is striking.
“pseudodominant EPP” came into use for patients with a FECH mutation trans to a hypomorphic IVS3-48C allele, to distinguish them from those with autosomal recessive EPP who are hetero-or homoallelic for FECH mutations. The frequency of the hypomorphic IVS3-48C allele differs widely between ethnic groups, ranging from 67.8% in Japanese to <1% in West Africans, and in general correlates with prevalence of clinical EPP among individuals with a mutant FECH allele in the different populations.176 When a low-expression allele is not found, a dominant-negative effect of FECH mutants may be operative.200 Because the enzyme activity is restricted to its homodimers,201 a mutant monomer may generate nonfunctional homodimers and impaired or unstable heterodimers, resulting in residual enzyme activity of <50% of normal.202, 203 Thus, the ultimate effect of a specific mutation in the FECH gene depends on how it affects the integrity of the protein and whether the nonmutated allele is expressed at a lower level to reduce FECH activity to below a threshold of around 30% of normal as found in clinically overt EPP.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


