T Lymphocytes and Natural Killer Cells
Frixos Paraskevas
MORPHOLOGY
The T lymphocyte under routine staining procedures (Giemsa or Wright) is 5 to 8 µm in diameter, with a high nucleocytoplasmic ratio. The nucleus is purple with densely packed chromatin, and the cytoplasm forms a narrow light-blue rim.
By transmission electron microscopy, the nucleus shows shallow indentations with dense heterochromatin along the nuclear membrane and euchromatin occupying most of the remaining nuclear surface. One or two nucleoli are visible (Fig. 13.1). The cytoplasm shows a few organelles, such as mitochondria and a small Golgi apparatus. By scanning electron microscopy, the surface of the T lymphocytes is either smooth or shows short microvilli, depending on the method of preparation as well as the state of activation.1,2
Some T lymphocytes present in normal subjects are characterized by a highly indented nucleus and are known as cerebriform mononuclear cells.3 These cells are not detected in T-lymphocyte-depleted fractions and constitute approximately 3% to 4% of the unfractionated T lymphocytes. They possess scanty cytoplasm, and the degree of their nuclear indentation is expressed as a nuclear contour index (nuclear perimeter/area).4 Their structural similarity to the cells present in cutaneous T-cell lymphomas suggests that they represent the normal equivalent of Sézary cells, which are derived from T lymphocytes.
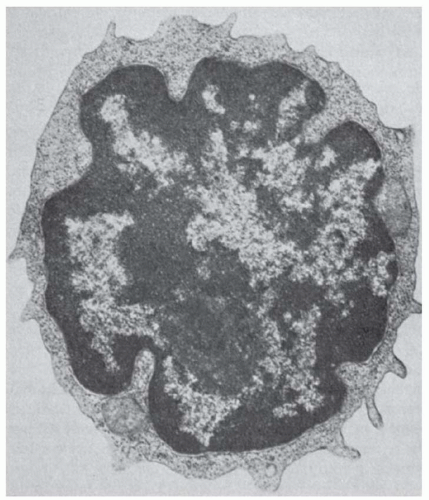 FIGURE 13.1. Ultrastructure of T-cells. The normal T-cell has a small rim of cytoplasm with only a few organelles. Nucleus shows dense heterochromatin. (From Zucker-Franklin D, Greaves MF, Grossi CE. Atlas of blood cells, 2nded. Philadelphia: Lea & Febiger, 1988, with permission.) |
ONTOGENY OF THYMIC MICROENVIRONMENT
The thymic anlage develops from epithelial structures of the third branchial complex (Fig. 13.2). Neural crest cells (Hoxa3-positive) invade the epithelial cluster to form the thymic rudiment. Several studies support the view that the cortical and medullary epithelia originate from a common precursor, and, in the early stages, the epithelium co-expresses markers that are later segregated to the cortical or medullary compartments. After completion of thymic organogenesis, the cortical epithelia are cytokeratin-8+, whereas medullary epithelia are cytokeratin-5+,5 except for a small subpopulation in the corticomedullary junction, which is cytokeratin 5+/8+.5 Furthermore, neoplastic human thymomas often express both cortical and medullary epithelial markers.6
The best evidence of the origin of thymic epithelia from a common progenitor is the identification by monoclonal antibodies of a cell that generates in vivo the thymic microenvironment.7,8 The monoclonal antibody MTS24 detects a glycoprotein with mucinlike characteristics and a peptide backbone.7 The antigen is detected during the early embryonic stages in the anterior
endodermal epithelium, the pharyngeal endoderm, and a portion of intermediate mesoderm, which develops the urogenital epithelium. The MTS24+ cells are positive for major histocompatibility complex (MHC) class II and express cytokeratin-5 and cytokeratin-8, which are markers of the medullary and cortical epithelium, respectively. Highly purified MTS24 cells, inserted under the kidney capsule, are able to develop into a complete thymus and, furthermore, provide the complete milieu necessary for T-cell development.
endodermal epithelium, the pharyngeal endoderm, and a portion of intermediate mesoderm, which develops the urogenital epithelium. The MTS24+ cells are positive for major histocompatibility complex (MHC) class II and express cytokeratin-5 and cytokeratin-8, which are markers of the medullary and cortical epithelium, respectively. Highly purified MTS24 cells, inserted under the kidney capsule, are able to develop into a complete thymus and, furthermore, provide the complete milieu necessary for T-cell development.
 FIGURE 13.2. Morphogenesis of thymus. Elements from the third pharyngeal cleft form the epithelial rudiment, joined by mesenchymal cells from the neural crest. The common epithelial progenitor differentiates into cortical and medullary epithelium under the influence of the mesenchyme. The presence of endodermal cysts and tissue-specific “antigens” and “organoids” suggests some contribution from the endoderm. FGF, fibroblast growth factor; KGF, keratinocyte growth factor. |
Because primordial epithelial cells normally need the cooperation of mesenchymal cells for thymus organogenesis,9 it is conceivable that, in the case of the ectopically placed MTS24 cells, this is provided by comparable cellular elements from the kidney capsule. The MTS24 antibody completely blocks T-cell development in fetal thymic organ cultures, supporting the conclusion that the molecule recognized by the MTS24 antibody regulates normal epithelial function. During this early stage in the development of the thymus, the cell interactions are regulated by a number of transcription factors, such as HOXA3 and PAX1 which are part of cluster A on chromosome 7 and encode a DNA-binding transcription factor which may regulate gene expression, morphogenesis, and differentiation.10,11 An important step in our understanding of the genetic control of thymic development came from the study of the “nude” mouse and the cloning of the gene that confers the nude phenotype, designated Whn, for Winged h n,12 more recently renamed Foxn1. The Foxn1 gene encodes a transcription factor with a DNA-binding domain of the forkhead/winged helix class. The defect responsible for the nude phenotype is a single base pair deletion in the third exon of the Foxn1 gene. It results in hair loss, the arrest of the thymic epithelial cell expansion, and inability to attract the hematopoietic precursors into the epithelial rudiment.13,14 Formation of the epithelial primordium is not affected by the loss of Whn function, but subsequent differentiation of the primitive epithelial precursors into subcapsular, cortical, and medullary epithelial cells is arrested15 and the epithelial rudiment becomes cystic.
In normal development, the epithelial rudiment is invaded by mesenchymal cells from the neural crest,16 which stimulate epithelial progenitor differentiation into cortical and medullary subpopulations.8,17,18,19 These mesenchymal-epithelial interactions are mediated by growth factors and their receptors, such as the fibroblast growth factor 10 (FGF10) and its specific receptor, FGFR2IIIb, on thymic epithelial cells. Deficiency of either the factor or the receptor results in severe thymic hypoplasia.20,21 When immature lymphoid cells begin to arrive in the thymic rudiment, another wave of cellular interactions takes place between the developing lymphoid cells and the stromal epithelial cells. This second stage of thymic development establishes thymic microenvironments conducive to thymocyte differentiation and repertoire selection. Further differentiation and maturation are under the regulation of interactions with thymocytes. Experiments in animals and experiments of nature in humans (disease processes) have clearly shown the symbiotic relationship between epithelial cells and lymphocytes. Prothymocytes regulate differentiation of cortical epithelial cells,22 whereas mature thymocytes organize the medullary microenvironment.23,24,25
The epithelial cells in the cortex and medulla differ by ultrastructure, by immunophenotype, and by functional characteristics. Ultrastructurally, three subsets have been detected on the basis of cytoplasmic processes, secretory organelles, and desmosome connections.26 Type I cells, located in the cortex, have stellate cytoplasmic processes and make contact with their neighbors, forming a syncytium. Type II cells are found in the medulla, are voluminous with many secretory intracellular vesicles, and have short cytoplasmic processes. Type III cells are rare, may contain vacuoles (pseudocysts), and are located in the medulla.
Six clusters of thymic epithelial staining (CTES) have been identified: type 1 epithelium (CTES II)27 produces thymic hormones, type II (pale), type III (CTES III) of intermediate electron lucency, and type IV (dark with oval nuclei) are in the cortex proper. These cells extend fine cytoplasmic processes, establishing contacts with neighboring cells, and are connected by desmosomes forming a syncytium, the interstices of which are filled with thymocytes. Some tend to engulf up to 20 to 40 thymocytes in a lymphoepithelial cluster, known as nurse cells, detected in human thymus28,29 (Chapter 11, Fig. 11.9). The internalized thymocytes are located within caveolae lined by plasma membranes. The lack of penetration by certain dyes indicates that the nurse cells are not an artifact, are completely sealed from the rest of the thymus, and may play a role in T-cell selection. Their formation is not dependent on interactions of T-cell receptors (TCRs) with MHC because they are present in knockout mice deprived of TCR-αβ.30
A better understanding of the thymocyte-epithelial cell interdependence for survival came from experiments with a variety of TCR-transgenic mice.31 The final organization of the cortical and medullary epithelium depends on interactions of the TCR on the thymocytes with ligands on stromal cells. Thymocytes expressing a transgenic TCR that triggers strong positive selection, resulting only in maturation of CD4+/CD8– T-cells, lose the normal reticular pattern of cortical epithelial cells, and the epithelium in the medulla forms small scattered groups of cells surrounded by macrophages and dendritic cells.32
Several other transgenic models demonstrated that maintenance of a balance among positive selection (PS), negative selection (NS), and nonselection is necessary to preserve normal compartmentalization and architectural integrity of the thymic epithelia.33,34 It is the diversity of signals emanating from these physiologic processes within the thymus during thymocyte maturation that are critical for the maintenance of epithelial organization. These signals are generated from the endogenously rearranged TCRs, which regulate positive and NS. In addition to thymocytes, the thymic vasculature is also an important epithelial organizer.35 In Rag2-/- mice, the medullary epithelium forms cuffs around intermediate-sized vessels, particularly the post-capillary venules. This anatomic arrangement may have important functional implications. The medullary epithelial cells have been linked to NS and tolerance induction36,37 and 38 and are therefore strategically located around the post-capillary venules, where the concentration of autoantigens would be high. The possible functional importance of this peculiar anatomic arrangement of the medullary epithelium is also suggested from the detection of several molecules and structures considered to be tissue-specific within the medulla (i.e., parathyroid hormone, thyroglobulin, insulin, and even organized epithelial “organoids” with ultrastructural features of respiratory epithelium and thyroid follicles).39 The ectopic location of these molecules and structures is supported by the detection of the expression of the appropriate genes.39 It has also been postulated that, because these “ectopic” tissues within the medulla have their origin from primordial endoderm, the precursors of the epithelial cells in the medulla also may arise from pharyngeal endoderm. Further support of this view comes from the histologic appearance of medulla in athymic mice, in which further differentiation of these cells fails and the epithelium remains in the primordial condition of endodermal cysts.
Whereas epithelial development depends on FGF, its functional integrity is maintained by other growth factors contributed by the mesenchyme. The keratinocyte growth factor (KGF), a member of the FGF family, is a paracrine growth factor produced by mesenchymal cells. It acts on epithelial cells that express a splice variant of FGF (i.e., FGFR2IIIb). In fetal thymic organ cultures, exogenous KGF expands the medullary epithelium, and in Rag-/- animals deprived of thymocytes, which normally produce KGF, the administration of KGF restores the normal medullary epithelial architecture.40
The architectural integrity of the thymus is maintained throughout life, but eventually it atrophies with age.41 Changes
in signals between epithelial cells and thymocytes may determine thymic involution.42 Stat3 has been identified as an important signaling molecule between epithelial and mesenchymal cells in the thymic microenvironment.43 Stat3 gene disruption in mice results in severe thymic atrophy and enhanced susceptibility of the thymus to environmental stress, such as glucocorticoids or γ-irradiation.
in signals between epithelial cells and thymocytes may determine thymic involution.42 Stat3 has been identified as an important signaling molecule between epithelial and mesenchymal cells in the thymic microenvironment.43 Stat3 gene disruption in mice results in severe thymic atrophy and enhanced susceptibility of the thymus to environmental stress, such as glucocorticoids or γ-irradiation.
The extracellular matrix (ECM) is an important component of thymic stroma after the cells and certain interactions between differentiating thymocytes and thymic epithelial cells are mediated by ECM proteins that influence intrathymic migration events and thymocyte differentiation.44 The ECM consists primarily of fibers of collagens, reticulin, glycosaminoglycans, and glycoproteins, including laminin and fibronectin.
Heterotrimeric laminin molecules consist of at least 15 naturally occurring isoforms, which are formed by five α, three β, and three γ subunits. In the human thymus, laminin with α2-chains (LN-2/4) or α5-chains (LN-10/11) are detected in the subcapsular epithelium and blood vessels.45 The CD4–/CD8– (double negative) thymocytes are located in the subcapsular area by strong attachment to LN-10/11 through their α6β1 integrin. The CD4+/CD8+ (double positive) thymocytes, however, lose their capacity to adhere to LN-10/11 and move down to the cortex.
The interactions between thymocytes and stroma facilitate intrathymic migration and regulate positioning of the developing thymocytes to appropriate microenvironments during differentiation. The ECM proteins support the growth of thymocytes and epithelial cells and facilitate cell-cell interaction, especially migration of thymocytes in and out of nurse cells. Receptors for ECM proteins are highest in the double-negative precursors but gradually decrease with maturation.
T-CELL PROGENITORS
The T-cell progenitors in fetal life derive from the liver, whereas in adult life, they come from the bone marrow. The difference in stem cell origin has implications in subsequent lymphoid development, apparently as a result of precommitment or restriction of developmental options at the level of stem cell.46 Human thymus becomes fully differentiated by approximately the 15th week of gestation (i.e., approximately 7 to 8 weeks after colonization of the thymic rudiment). These early migrants contribute to the development of the thymic microenvironment.
Cells with the CD34+/CD38weak/CD90+ phenotype contain T-cell progenitors when they grow in fetal thymic organ cultures. In human adult bone marrow, CD34+/CD38–/HLA-DR+ stem cells have the potential to differentiate toward lymphoid and myeloid lineages.47 These cells are CD45RA+, lack Thy1 (CD90) antigen, and may represent an intermediate oligopotent stem cell with T-cell-reconstituting ability.48 By phenotype, there are three subpopulations in the bone marrow, which can differentiate to T-cells in the thymus: CD34+/CD2+, CD34+/CD7+, and CD34+/CD2+/CD7+ and all three are negative for CD3/CD4/CD8.49 The thymus is populated by hematopoietic multipotent progenitors from the bone marrow.50
Experiments with transgenic mice have shown that disruption of the zinc finger protein Zbtb1, resulted in a T(-)B+NK– SCID phenotype.51 In knockout mice for Zbtb1 protein differentiation of common lymphoid progenitors is inhibited and it is associated with expanded populations of bone marrow hematopoietic stem cells and multipotent and early lymphoid lineages51 corresponding to a common lymphoid progenitor subset.52 The most primitive thymocytes are Lin–, c-Kithigh, L-selectin+, TdT+, and RAG1–, a phenotype similar to progenitor population in the bone marrow which sustains the adult thymus.53
Recruitment involves the chemokines CCL21 (or SLC, secondary lymphoid tissue chemokine) and CCL25 produced by the fetal thymus, which attract CD4–/CD8–/CD25–/CD44+ thymocytes.54 At the site of entry P-selection of the thymic vascular endothelium binds arrests precursors expressing the P-selectin ligand (rolling). Chemokine CCL25 from endothelial cells activates precursors through the CCR9 receptor, and intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1(VCAM-1) of the endothelial cells bind the cells firmly through the integrins αLβ2 and α4β1, respectively.55 The multipotent T-cell progenitors entering the thymus trigger Notch activation, which is critical for T-cell development (see below) for later stages of TCR-dependent selection within the thymus as well as during peripheral T-cell differentiation.55 There is some overlap of regulatory programs between those required for T-cell specification and those needed for stem cell maintenance or self-renewal, such as Notch pathway, GATA3, IKZF1 (IKAROS), and SPI1 (PU.1).56 The genes that need to be turned on for a successful entrance in the early stage of T-cell development include RAG1 and RAG2 and CD3G and CD3E of the pre-TCR and TCR. Although certain options of differentiation are eliminated relatively early after T-cell lineage commitment, others such as differentiation into dendritic cells and natural killer (NK) cells remain through some stages of the T-cell development. The B-cell potential is lost early within the thymus probably as a result of Notch signaling. Additional factors, such as GATA3 (T-cell-specific among the hematopoietic cells), TCF3 (E2A), shared with B lineage and even non-T-cell-specific such as RUNX1 and IKZF1 are also used in T-cell development.
NOTCH AND T-CELL COMMITMENT
Signaling through the Notch receptor is a key factor for T-cell commitment.57,58,59 and 60 Notch plays a pivotal role in determining T-/B-lineage choice, and signaling through Notch drives commitment of lymphoid precursors to T lineage.61,62 and 63 Notch belongs to a family of conserved proteins that function as cell-surface receptors and direct regulators of gene transcription.64 It was first isolated as a gene involved in chromosomal translocations with the TRB@ gene in a subset of cases of human T-cell acute lymphoblastic leukemia. The extracellular domain of Notch contains a variable number of tandem epidermal growth-factor-like repeats and three Lin/Notch repeats, which function for ligand binding and Notch activation.65 The intracellular region contains six Cdc10/ankyrin repeats characteristic of protein-protein interactions and essential for signal transduction. The Notch protein initially is synthesized as a single-polypeptide chain, but, as a result of proteolytic processing, it is split into two parts. The extracellular region is separated and forms a noncovalent heterodimer with the remaining portion consisting of the transmembrane and the cytoplasmic regions.
Ligands for Notch are Delta, Serrate, and several other molecules corresponding to these two classes. In general, those homologous to Delta are referred to as Delta, and those homologous to Serrate are called Serrate or Jagged.65 These ligands are transmembrane proteins with an extracellular domain with a variable number of epidermal growth-factor-like repeats and the unique domain for this family, the DSL (Delta/Serrate Lag-2) domain, which mediates binding to Notch and activation. Which of the two groups of ligands is important for lineage commitment remains controversial.66 In cells of the immune system, there are two Notch receptors, NOTCH1 and NOTCH2, and four signals.67 The pleiotropic signaling by Notch regulates differentiation, proliferation, and cell death, but it is not yet clear which function most precisely determines cell fate and ultimately directs T-cell commitment.68 With Notch inactivation, the double-negative T-cells diminish in the thymus, whereas B-cell precursors increase, probably from a more efficient production of B-cells within the thymus.69 On the other hand, transgenic expression of Notch in the bone marrow permits the accumulation of CD4+/CD8+ T-cells. The NOTCH1 functional role seems to
be in developmental specification, driving T-cell precursors at the expense of B-cell precursors and perhaps directing the choice of a common precursor between these two fates. Notch is also needed for later stages for TRB@ gene rearrangements, PS, and CD4/CD8 lineage choices.56
be in developmental specification, driving T-cell precursors at the expense of B-cell precursors and perhaps directing the choice of a common precursor between these two fates. Notch is also needed for later stages for TRB@ gene rearrangements, PS, and CD4/CD8 lineage choices.56
PHENOTYPIC DIFFERENTIATION
The first migrants from the bone marrow to the thymus settle in the corticomedullary junction and are large dividing cells expressing CD34+/CD45RA+, CD2+, and CD7+, with the TCR genes in the germline configuration (Fig. 13.3). The new immigrants potentially are able to differentiate to other lineages, such as NK-cells, dendritic cells, and monocytes. The CD34+ cells then co-express other markers such as CD38 and CD71 (transferrin receptor) associated with proliferating cells, and a portion of them are CD10+. They are subdivided into three categories, double negative-1 (DN-1), DN-2, and DN-3, depending on the expression of CD44 and CD25.70,71 Recruitment involves interactions between P-selectin expressed by thymic endothelium and P-selectin glycoprotein ligand-1 detected on the bone marrow progenitor cells.72 Upon arrival they migrate to the subcapsular cortex (DN-3 cells), where they acquire expression of CD1A and CD4 and CD8, and as a result become double positive (DP). In the cortex they are submitted to PS (vide infra) and those considered “useful” return back to the medulla, and they are separated into two distinct phenotypes, CD4+ and CD8+ single-positive (SP) cells. These migrations across the thymic parenchyma are mediated by several chemokine receptors, CXCR4, CCR7, and CCR9. Adherence of thymocytes to epithelial cells is also mediated by CD2 and the lymphocyte function molecule-3 (LFA3, CD58), ICAM, and LFA1. The SP thymocytes are submitted to another screening in the medulla. Those with high-affinity TCR against self-antigens are deleted (NS). After this checkpoint, the surviving thymocytes exit the thymus for the secondary lymphoid organs. Emigration of thymocytes requires the sphingosine-1-phosphate receptor 1 (S1P-1).
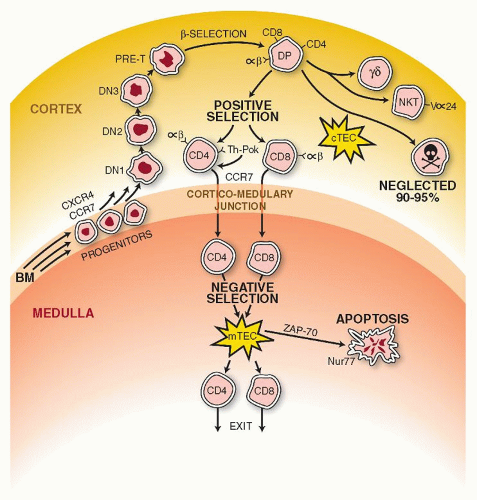 FIGURE 13.3. The first migrants from the bone marrow arrive and settle in the corticomedullary junction. They lack expression of CD4 and CD8 and are known as double negatives (DN). These new immigrants begin to move to the outer cortex and, depending on certain markers, are distinguished in three stages: DN1, DN2, and DN3. The migration is supported through interactions between P-selectin (thymic epithelium) and its ligand (progenitor cells). In the outer cortex they become double positive (DP) (i.e., CD4+/CD8+), and at the DN3 stage, undergo gene rearrangements with expression of the T-cell receptor (TCR)-β gene (β selection). Signals provided by the CCR-7 chemokine receptor guide the positively selected thymocytes that have already separated into the CD4 or CD8 lineages back to the medulla. A final screening for the “affinity” of their TCR binding to autoantigens takes place in the medulla (NS). It is triggered by contacts of their TCR with tissue-restricted self-antigens (TRAs) on medullary thymic endothelial cells (mTECs). Promiscuous gene expression (pGE), for autoantigens, regulated by the AIRE gene (see text for details), controls central tolerance. The thymic medullary epithelium ultimately allows survival of the “useful,” and it ignores the “useless” and “destroys” the harmful128. Exit of mature T-cells from the thymus is regulated by the G-protein-coupled receptor sphingosine-1-phosphate receptor 1 (S1P-1) as well as the very late antigen (VLA-) and lymphotoxin-β receptor. |
In the process of differentiation, CD34 is progressively lost, whereas the intensity of CD7 decreases. Myeloid and NK-cells have been detected in various in vitro systems arising from thymocytes, 73,74 and the thymic microenvironment is able to support myeloid differentiation.
Evidence of direct cellular communication between various thymic cells was provided by the demonstration of the existence of gap junctions formed by connexin 43 between two epithelial cells or between epithelial cells and thymocytes.75
Of the cytokines that have been implicated in T-cell differentiation, interleukin-7 (IL7) is essential.76 It is produced constitutively by epithelial cells, and it induces proliferation of DN thymocytes77 or maintains their viability.78 CD34+ thymocytes cultured with IL7 start to express CD8 and CD4 but remain CD3 and TCR negative, indicating that other stimuli from stromal cells are essential for generating CD3+/CD4+/CD8+ cells. IL7 also induces Tcrb gene rearrangements.79 Mice genetically deficient in IL7 receptor have a profound reduction of T and B lymphocytes, and thymocyte development is blocked at a very early stage before the induction of CD25 and Tcrb gene rearrangements.80 IL7 regulation is tightly coordinated during T-cell maturation within the thymus for thymocyte survival, as IL7 is essential for post-selection expansion of positively selected thymocytes.81
Other cytokines, such as IL1, IL2, and IL4, have also been shown to play some role in thymocyte differentiation.76 The thymus is continuously colonized by hematopoietic progenitors that have the genes for the Tcr in germline configuration. Approximately 100 to 1,000 such progenitors enter the thymus daily, and it takes approximately 3 weeks to undergo complete differentiation to mature functional and self-tolerant T-cells.
The biologic processes within the thymus are highly complex, but for a better understanding, we divide them, somewhat arbitrarily, into three areas: (a) lineage determination i.e., TCR-αβ versus TCR-γδ, (b) separation of the two main T-cell subsets, i.e., CD4 versus CD8, and (c) selection for survival of those cells with a TCR able to recognize foreign antigens, i.e., PS and elimination of those possessing autoreactive configurations, i.e., NS. Understanding of these events will be facilitated by a prior description of the genes encoding the Tcrs.
T-CELL RECEPTOR GENES
Knowledge of the structure and patterns of expression of the various TCR genes is essential to our understanding of antigen recognition by T-cells. The TCR gene, “a needle in the haystack,” was isolated by the technique of subtractive hybridization.82 Four human and murine TCR genes have been identified: α (TRA@, Tcra),83 β (TRB@, Tcrb),84 γ (TRG@, Tcrg), and δ (TRD@, Tcrd)85,86 (Fig. 13.4).
The TRA@ gene is located on chromosome 14 (bands q11-12),87 as is the immunoglobulin (Ig) gene (band q32). Rearrangement involving the region of the chromosome containing TRA@ has been detected in patients with T-cell malignancies.88,89 The TRD@ gene is located on chromosome 14 within the TRA@ gene.90 The TRB@ and TRG@ genes are located on chromosome 7.91,92 Translocations and inversions of chromosomes 14 and 7 are
often seen in association with ataxia telangiectasia, probably involving fragile sites that normally are used during the TCR gene rearrangements.
often seen in association with ataxia telangiectasia, probably involving fragile sites that normally are used during the TCR gene rearrangements.
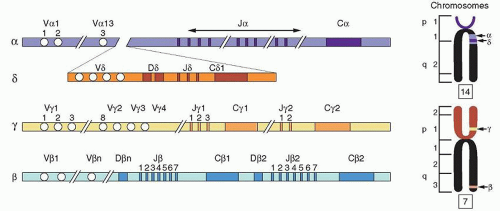 FIGURE 13.4. Organization of the genes for human T-cell receptor chains (see text for details). (Adapted from Kronenberg M, Siu G, Hood LE, et al. The molecular genetics of the T-cell antigen receptor and T-cell antigen recognition. Annu Rev Immunol 1986;4:529-591; and Raulet DH. The structure, function, and molecular genetics of the γ/δ T-cell receptor. Annu Rev Immunol 1989;7:175-207.) |
All TCR genes display an overall organization similar to that of the Ig genes. They are composed of variable (V) and constant (C) genes. The V gene is made of three segments (V, J, and D) in the TRB@ and TRD@ genes, but only two segments (V and J) in the TRA@ and TRG@ genes.86 Each V gene family is divided into subfamilies like the Ig V genes, on the basis of sequence similarity (more than 75%). In humans, the TCR gene group consists of one C-α gene,93 two C-β genes,94 two C-γ genes, and one C-δ gene. With the exception of the C-γ gene, which is composed of three exons, all of the genes have four exons. The TRD@ locus is located between the C-α and J-α gene segments. Diversity of the TCR genes is generated through rearrangements, with each of the V and J segments and the D segment, in the case of TRB@ and TRD@ genes, forming the complete V gene. The TCR genes, however, contain a large number of J segments as compared to the Ig genes.
Associated with the C-α gene are approximately 50 Vα and 61 Jα segments that spread over 100 kb of DNA. The β-chain (TRB@) gene complex is located on chromosome six, spans 600 kb95 and incorporates 57 V-β segments. Each of the two C-β genes possesses a set of J-β segments and one D-β segment. The V-β segments are all located upstream from the two clusters of C-β genes. During rearrangements, when a V-β segment forms a VDJ complex, the transcript is committed to using the same C-β segment genes. The TRG@ gene locus contains two C-γ genes, each associated with its own set of J-γ segments. There are approximately 14 V-γ segments, all located upstream separately from the two Cγ-Jγ clusters. The C-δ gene complex lies between the V and J segments of the TRA@ gene complex and contains eight V-δ, three D-δ, and three J-δ segments. The TCR gene segments, as are the Ig gene segments, are flanked by heptamer-spacer-nonamer sequences, which serve as recognition sites for the recombinase, the enzyme that initiates V(D)J recombination which is regulated by the RAG1 and RAG2 genes for both T- and B-cells. The RAG1/RAG2 proteins act in a lineage-specific manner; i.e., Ig genes are assembled only in B-cells and TCR genes only in T-cells. The recombinations are also developmentally regulated. For T-cells the β genes are recombined before TRA@ genes and for B-cells the IGH@ gene is recombined before IGL@. These differences are explained by differential accessibility of V genes during development. Accessibility for V(D)J recombination is usually accompanied by germline transcription at antigen receptor loci.96 Two mutually exclusive T-cell lineages have been identified on the basis of TCR: αβ and γδ.
Transcription increases the recombination of J (α) segments located within several kilobases of a promoter and prevents the activation of downstream promoters.97 The mechanism of V(D)J recombination has been described in detail in Chapter 12.
The mechanisms generating diversity are combinatorial associations of different V, D, and J segments and combinatorial pairing of TCR protein. However in contrast to the Ig genes, fewer V segments are available to the TCR genes. The diversity of the TCR is mainly junctional, i.e., a result of additions of nucleotides at the DNA cleavage site. Additions that depend on the template are known as P nucleotides, whereas random additions are called N nucleotides, added by terminal deoxynucleotidyl transferase (TdT) (see Chapter 12). TdT is not expressed during fetal life and in TdT knockout mice, the T-cell repertoire is of fetal type (i.e., less diverse). TdT is expressed only in immature lymphocytes and is responsible for the transition from fetal to adult repertoire by contributing significantly to the lymphocyte antigen receptor repertoire. A striking difference between Ig and TCR is the lack of somatic hypermutations, which is very important in generating high-affinity antibodies in the germinal centers (Chapter 11). Assembly of V segments of TRB@, TRG@, and TRD@ genes occurs during the DN stage. If the β-chain rearranges successfully, it forms a heterodimer with the pre-Tα (PTA) chain and differentiates along the T-αβ lineage entering the DP phenotype stage.
TCR gene rearrangements occur in two discrete stages of thymocyte development: first during the DN stage with the TRD@ gene rearranging first, followed by the TRG@ and TRB@ genes and the TRA@ gene rearranging during the DP stage. During the TRB@ gene rearrangement, a D-β segment joins a J-β segment and as with the IGH@ gene rearrangement, a V-β joins the Dβ-Jβ complex. If the rearrangement is not productive the cell may have one more chance at rearranging a second C-β cluster inasmuch as there are two C-β segments each with its own D and J segments.
PRE-T-CELL RECEPTOR AND T-CELL-αβ, T-CELL-γδ LINEAGES
The decision in choosing a TCR-γδ versus a TCR-αβ lineage is made by any one of three proposed models. According to one mechanism each lineage starts from a separate precursor
(stochastic or independent pathway), and a second model proposes that there is a common precursor for both lineages (instructive pathway). The third model (competitive pathway) postulates that rearrangements for the genes of the lineages start concurrently and those finishing successfully first determine the fate of the cell. Some evidence appears to support a variant of the stochastic model.96 It is agreed that the TRD@ gene rearrangement occurs in thymocytes that can adopt either one of the γδ or αβ fates, but activation of the TRA@ gene rearrangement during the DP stage seals the fate of the T-cell for the αβ lineage.
(stochastic or independent pathway), and a second model proposes that there is a common precursor for both lineages (instructive pathway). The third model (competitive pathway) postulates that rearrangements for the genes of the lineages start concurrently and those finishing successfully first determine the fate of the cell. Some evidence appears to support a variant of the stochastic model.96 It is agreed that the TRD@ gene rearrangement occurs in thymocytes that can adopt either one of the γδ or αβ fates, but activation of the TRA@ gene rearrangement during the DP stage seals the fate of the T-cell for the αβ lineage.
Determination of lineage is regulated by transcription factors that act on promoters and enhancers and make appropriate genes accessible to the recombinase.97,98 Another regulatory mechanism involves the pre-TCR. At this stage of T-cell development, the TCR-β chain forms a heterodimer with the PTAchain and the complex is referred to as pre-TCR.99 The PTA is a 33-kd type I transmembrane protein of the Ig superfamily, with a single Ig-like domain. Two cysteines form the intrachain disulfide bond, whereas a third cysteine just above the transmembrane region forms the disulfide bridge with the TCR-β chain.100 The human PTA gene is located on the short arm of chromosome 6, in the vicinity of the HLA locus. The PTA chain is not essential for CD3 expression, but has a major role in TCR-αβ versus TCR-γδ commitment. It generates large numbers of CD4+/CD8+ T-cells with productive TCR-β rearrangements and directs these cells to the TCR-αβ lineage.101 At the DN-3 stage, thymocytes undergo extensive DNA rearrangements at the TRB@, TRG@, and TRD@ gene loci and make a choice for selection, between the TCR-αβ and TCR-γ receptors. Expression of the TCR-β chain in combination with the pre-TCR-α chain results in a process known as β selection, which leads to rearrangements of the TRA@ genes and formation of the complete TCR-αβ. If the TRD@ or TRG@ genes are rearranged, the cells follow the TCR-γδ lineage (γδ selection).102,103 and 104
The selection for TCR-β is initiated at a stage phenotypically characterized by expression of CD4+/CD8α+β–.105 The productive V-β gene rearrangement in one allele prevents rearrangements of the second allele, a process called allelic exclusion that is regulated by the pre-TCR. Signaling by the pre-TCR requires the CD-3 chains γ, ε, ζ, and ε, and the LCK kinase,106 which is associated with the CD3-ε and –γ chains and is indispensable for the pre-TCR function. Other functions mediated by pre-TCR signaling are cell survival and phenotypic changes. Pre-TCR promotes thymocyte survival, whereas signaling by TCR induces apoptosis.107,108 Because pre-TCR is expressed on DN T-cells, the antiapoptotic function of pre-TCR is crucial for their survival and differentiation of DN T-cells into the DP stage when the TCR is needed for positive or NS.
Both receptors follow initially similar signaling pathways, such as tyrosine phosphorylation and so forth, but in the apoptotic pathway, they diverge at the level of Fas ligand (FasL) induction, which requires induction of Nur-77 and transcription factors from the nuclear factor of activated T-cells (NFAT). These factors can be induced only by the TCR-αβ but not by the pre-TCR. The PTA possesses a palmitoyl moiety that spontaneously targets the PTA chain to the cell membrane (lipid rafts).109 This may offset the requirements for a ligand because the pre-TCR from such a location is able to signal constitutively. Signaling through pre-TCR results in expression of certain transcription factors required for the differentiation of precursors to αβ T-cells, i.e., β selection (see above). Some of these factors are the E-proteins of the basic helixloop-helix transcription activators required for expression of CD4, TCR-α and TCR-β chains and so forth.110
CD4/CD8 LINEAGE COMMITMENT
The DP thymocyte (CD4+/CD8+) differentiates into two phenotypically and functionally distinct lineages of αβ T-cells: CD4+ and CD8+. The TCR of the CD4+ cells interacts with peptides bound to class II MHC molecules, whereas the TCR of the CD8+ cells recognizes peptide-class I MHC complexes. The CD4 and CD8 proteins are not clonally distributed and are known as coreceptors because they recognize the same ligands as the TCR.111 The mechanism by which the separation of the lineages from DP thymocytes is achieved remains unresolved.112,113
According to one theory, known as instructive, thymocytes carrying an MHC class I restricted TCR differentiate to CD8+ lineage, whereas engagement of TCR with class II MHC induces commitment to CD4. An alternative model, known as stochastic (or selective), accepts that the DP thymocytes are already committed randomly to a lineage; they make a choice that is unrelated to the MHC specificity of their TCR. Data collected from a variety of approaches do not agree with any particular mechanism in lineage commitment. The instructive model proposes that the CD4 or CD8 transduces differentiation-specific signals, but no lineage-specific signals have ever been identified.
It is also possible that the cytoplasmic region of CD4 directs CD4 lineage commitment. A chimeric construct, for example, made of the cytoplasmic region of CD4 and the extracellular and transmembrane region of CD8a supported the development of cells with the CD4+/CD8a– phenotype but with a class I restricted TCR.114 CD4 is preferentially associated with its cytoplasmic tail with the tyrosine kinase LCK. Therefore, CD4 is likely to deliver stronger signals than the coengagement of TCR and CD8.115,116
The strength of signal model suggests that strong signals dictated by the frequency of the available ligand induce CD4 differentiation, whereas weak signals induce CD8. Another aspect of signaling that was evaluated was the duration of signals as an important parameter. When the interaction is limited to a few hours, the cells become CD8+, but exposure for longer periods of time results in CD4+ lineage commitment.117 Accordingly, TCR of a DP thymocyte initiates down-regulation of CD8 and produces a CD4+/CD8– intermediate cell. At this stage, the duration of signaling determines the final outcome. That is, short signaling produces CD8+ T-cells, whereas persistence of signaling in the CD4+/CD8– intermediate cell causes CD4+ differentiation.118 Variation of LCK function seems to be the single most important parameter in CD4/CD8 lineage decision. Constitutively active LCK promotes CD4 differentiation, even in the presence of class I MHC-restricted TCR. When, on the other hand, LCK is catalytically inactive, all thymocytes including those with class II MHC-restricted TCR, become CD8+.119,120 In the absence of LCK, cross-linking of CD3 induces CD8 differentiation.121 The LCK-dependent regulation of lineage commitment is not only phenotypic, but also functional, because the class II MHC-restricted CD8+ cells behave as killer cells, whereas the class I restricted CD4+ cells up-regulate CD40 ligand, a function characteristic of helper T-cells.119 Signaling initiating from LCK is channelled through the Ras-Erk pathway.122
The various experimental approaches used in these studies make it clear that the signals required for CD4 differentiation are promiscuous, which is believed to indicate that CD-4 differentiation is a default pathway.123 The multiplicity of the models entertained and the ambiguity of some of the results are also a testimony that the precise mechanism of T-cell lineage commitment remains still elusive and certainly complex.113,124
More recently a new transcription factor Th-POK (T-helper-inducing POK factor) was found to be necessary and sufficient for CD4 lineage commitment and absence of Th-POK results in the development of CD8+ T-cells.125 The existence of this factor was suspected as a result of studies of the spontaneous recessive mutant mouse with helper deficient cells (i.e., CD4+ T-cells).126 Th-POK belongs to the POK family of transcription factors characterized by two motifs, a regulatory POZ/BTB domain (for interaction with other transcription factors) and a Zn finger DNA-binding domain.
In summary, LCK is a key regulator, but other signals originating from the TCR in the absence of a coreceptor or in the
absence of a significant recruitment of LCK provide sufficient SRC activity for a response. Such signals depend on the nature of the peptides involved in positive versus NS, which is linked to lineage selection.121 Although the prevailing opinion accepts that lineage commitment and the selection of thymocytes with useful TCRs are linked, some of the models proposed point to the opposite.127 The latest view that CD4/CD8 lineage commitment is transcriptionally regulated will put an end to this question hotly debated for a long time.
absence of a significant recruitment of LCK provide sufficient SRC activity for a response. Such signals depend on the nature of the peptides involved in positive versus NS, which is linked to lineage selection.121 Although the prevailing opinion accepts that lineage commitment and the selection of thymocytes with useful TCRs are linked, some of the models proposed point to the opposite.127 The latest view that CD4/CD8 lineage commitment is transcriptionally regulated will put an end to this question hotly debated for a long time.
POSITIVE AND NEGATIVE SELECTION
The random nature of rearrangements of TCR genes generates specificities directed not only against foreign antigens, but also against self-antigens. Self-reactive T-cells are harmful if they have the opportunity of exiting the thymus. Mechanisms have therefore been developed that allow the thymus to “select the useful, neglect the useless and destroy the harmful.”128 The time for selection appears to begin at the DP stage when the TCR is expressed at low levels.129,130,131 and 132 The TCR of the thymocytes recognizes self-peptides presented by the MHC molecules. It has been widely accepted that the options for each DP thymocyte are determined by some fine qualities and properties of the TCR and MHC-peptide (pMHC) ligand interaction. If the affinity of interaction is “weak,” the cell is positively selected, whereas a T-cell with high affinity is negatively selected.133
Cells with TCRs that cannot bind with sufficient avidity are neglected and die by apoptosis, whereas in the positively selected cell the expression of RAG, which encodes the proteins regulating V(D)J recombination and the PTCRA (pre-T-cell receptor) gene are turned off and expression of CD4 and CD8 are partially down-regulated. At this stage the cell moves from the cortex to the medulla, one of the coreceptors is re-expressed and the cell lineage is defined.122,134 Signaling by LCK regulates positive and NS and in its absence the selection mechanisms are compromised.135 LCK interacts with the coreceptors CD4 and CD8 through two cysteines in the cytoplasmic region of each co-receptor and two cysteines in LCK.136
The binding of TCR to the ligand induces phosphorylation of the three immunoreceptor tyrosine-based activation motifs (ITAMs) of the ζ-chain and the single ITAM in each of the CD3 ε-, δ-, and γ-chains. The ζ-chain is constitutively associated in thymocytes with the ZAP70 kinase, suggesting that a low level of activation takes place continuously.137 The fate of the immature thymocyte depends on the type of APC that it encounters in the various histologic compartments of the thymus. The cortical thymic epithelial cells usually mediate PS,138,139 and the medullary hematopoietic cells mediate NS,140 although under certain circumstances they may also contribute to PS.141 Interactions among thymocytes may also support PS if the selecting thymocyte carries MHC class II molecules.142 In this case the α-chain of the TCR, and particularly the highly diverse CDR3 loop of the α-chain, plays an important regulatory role.143 Signals provided by the CCR7 chemokine receptor144 guide the positively selected thymocyte to the medulla. The CCR7 signals provide guidance for the selected thymocyte through the corticomedullary maze, in preparation of the central tolerance in the medulla prior to their exit to the periphery.144 An important selective force in these cellular interactions is the strength of the interaction, which determines the outcome. Strong interactions lead to deletion, whereas intermediate strength induces PS. This may explain the increase of PS following blocking of CD28/B7 interactions.145 Removal of strong self-reactive cells is the sole purpose of the NS. Such clones potentially may initiate autoimmunity if they exit to the periphery. An estimated 2,000 to 3,000 tissue-specific antigens are expressed in human or murine medullary thymic endothelial cells (mTECs),146,147 i.e., 5% to 10% of the known mouse genes, in addition to their normal tissue expression. About 500 of these genes may be AIRE (autoimmune regulator) -dependent. Using hen egg lysozyme as reporter for the insulin promoter, it was shown that the expression of the antigen in the thymic stroma is necessary and sufficient for deletion.148
Expression of tissue-restricted self-antigens (TRAs) by mTECs is the result of promiscuous gene expression (pGE) by the mTECs.146 Some of the genes are expressed by both mTECs and cortical thymic endothelial cells (cTECs; pool 1), whereas others are expressed only by mTECs (pool 2), and finally others are expressed only in the more mature mTECs (i.e., strongly positive for MHC class II; pool 3). Depending on the stage of differentiation of mTECs, two models have been proposed for pGE: (a) in the developmental, or progressive restriction model, pGE is detected in immature and perhaps pluripotent progenitor cells and (b) in the terminal differentiation model, pGE is mutated in mTECs of a mature phenotype.149 The pGE is regulated by AIRE, the gene mutated in the rare autoimmune disorder known as autoimmune polyglandular syndrome type 1 (APS-1).150
AIRE has a nuclear localization signal and several potential DNA-binding and protein interaction domains. In cooperation with transcription factor CREB-binding protein it transactivates the transcription of other genes, although it is still a mystery how as a single molecule it controls the transcription of such an array of genes.
The pGE has been conserved across species barriers during evolution as a study recently with human pure populations of thymic cTECs and mTECs has shown.151 The AIRE gene showed the highest enrichment in mTECs and the promiscuously over-expressed genes are those remarkably well conserved among species. A set of 443 genes have been detected overexpressed in mTECs (comparable to 555 in mouse). This is an underestimate as most were detected by polymerase chain reaction. These genes show no preference for any chromosome. Another interesting aspect is the finding of clustering of nonhomologous genes. This clustering has been suggested to result from the juxtaposition during evolution of genes involved in the formation of a particular tissue. Because the promiscuously expressed genes do not all share such a function, the clustering is the result of epigenetic mechanisms of regulation. They become accessible to mTECs as a result of belonging to the same “gene neighborhood.” pGE not only is sufficient for self-tolerance, but also likely has been essential for survival of the species inasmuch as infertility as a result of gonad-specific autoimmunity is highly prevalent in APS-1152 and in Aire-/- mice.153 In addition to genetic control, self-tolerance may also be regulated by epigenetic mechanisms. The lymphotoxin β receptor (LTβR) directs the three-dimensional organization of mTECs,154 and TRAF6, a cytoplasmic adaptor molecule, which does not bind to LTβR, was still shown to direct mTEC development.155,156
Self-tolerance is mediated primarily by NS or clonal deletion,157 but some of the self-reactive T-cells are submitted to nondeletional central tolerance and give rise to immunosuppressive CD4+ T-cells, also known as natural regulatory T-cells (T reg) cells.158 T reg cells are T-cells with medium to high affinity for self-antigens. However, they escape NS and are positively selected by a subset of dendritic cells that have been educated by a thymic stromal lymphopoietin produced by Hassall corpuscles.159 Other evidence, on the other hand, indicates that CD4+ T reg cells are positively selected by thymic epithelial cells expressing self-antigens but not by bone-marrow-derived dendritic cells.160 ZAP70 is indispensable for positive and NS because both processes are abrogated in ZAP70-deficient mice.161
The SRC-like adaptor protein (SLAP) down-regulates the TCR expression during the DP stage of development and rescues T-cell development in the absence of ZAP70. Overall, SLAP acts as a
negative regulator and probably “marks” activated receptors for retention and degradation.162 The main downstream signaling pathway for PS is the Ras/map kinase (MAPK) cascade.163 TCR signaling alone is not sufficient to induce selection for survival or death.164 CD2 expression exerts a strong influence on the TCR repertoire. In the absence of CD2, the thymocytes with high affinity for peptide MHC escape NS.165 CD2 also influences pre-TCR function in the usage of Vα genes, which is substantially altered in CD2-deficient mice.
negative regulator and probably “marks” activated receptors for retention and degradation.162 The main downstream signaling pathway for PS is the Ras/map kinase (MAPK) cascade.163 TCR signaling alone is not sufficient to induce selection for survival or death.164 CD2 expression exerts a strong influence on the TCR repertoire. In the absence of CD2, the thymocytes with high affinity for peptide MHC escape NS.165 CD2 also influences pre-TCR function in the usage of Vα genes, which is substantially altered in CD2-deficient mice.
The costimulatory interaction of CD40/CD40L is a master regulator of NS, usually acting in the regulation of the ligands of other costimulatory molecules, such as CD80 and CD86.166 CD40 may also induce other co-stimuli required for thymocyte deletion, such as CD54 (ICAM-1), FASL, or tumor necrosis factor (TNF).167,168 and 169 These molecules could regulate NS separately or in combination with CD5 and CD28. The CD28 costimulatory molecule engaged with TCR signals thymocytes to undergo apoptosis or maturation, depending on the intensity of costimulation.170
Stimulation of maturation of DP thymocytes follows activation of the extracellular signal-regulated kinase ERK/MAPK pathway and up-regulation of the antiapoptotic protein BCL2. Apoptosis is triggered with the expression of the Nur77 family of transcription factors and occurs only if TCR engagement is accompanied by costimulation. Ca2+ fluxes in mature T lymphocytes regulate proliferation, differentiation, and survival. Some of the functions of Ca2+ are mediated by the Ca2+-dependent phosphatase calcineurin. The function of calcineurin is disrupted by cyclosporin A and FK506. Both of these substances form complexes with cellular proteins, termed immunophilins, that bind and sequester calcineurin. Cyclosporin A and FK-506 block the initial steps of PS.171,172 On the other hand, intracellular chelators reduce deletion,173 which suggests that interference of Ca2+ fluxes and function exerts several effects on the selection process.
All signaling pathways end up targeting regulation of transcription factors, and various studies have examined the roles of NFAT and nuclear factor-κB (NF-κB), but only members of the Nur-77 family have been clearly shown to be involved in the process of selection, with a major role in apoptosis of massive numbers of thymocytes.174 It has been estimated that 90% to 95% of thymocytes die inside the thymus, and the appearance of very large numbers of dead cells on histologic sections is the reason that histopathologists called the thymus the “graveyard” of T-cells. The timing of positive versus NS has been difficult to define. NS was believed to occur relatively late because the TCR affinity increases with time, and it is the high affinity of TCR that triggers NS.
THE CELLS AND MOLECULES IN THE SELECTION OF THYMOCYTES
Hematopoietic components of the thymic stroma are radiosensitive, whereas the epithelial components are radioresistant. These differences allowed the construction of chimeric thymuses (i.e., thymuses composed of epithelial and hematopoietic elements with a different MHC background). These experiments indicated that the thymic cortical epithelium is responsible for PS.175 The hematopoietic stromal cells, on the other hand, are potent inducers of NS. The cortical epithelial cells have weak MHC expression and the developing DP thymocytes at that stage have low TCR expression; as a result, the interactions are only of low avidity, required by positive. Similarly, the hematopoietic cells in the corticomedullary junction express MHC molecules at higher density, and the T-cells have up-regulated their TCRs. As a result, the interactions in this anatomic location are appropriate for NS. The concentration of the peptide may also play a role in the selection process. thymocyte bearing a TCR specific for class-I-peptide complex is positively selected if the peptide it carries is in a low concentration and is killed if the peptide concentration is high.176 However, the weak interactions required for PS and the strong interactions operating in NS can also be provided by the half-life of the pMHC complex (i.e., short half-life for positive and long half-life for NS).
The original question of affinity versus concentration was better answered with manipulation of the pMHC. PS is driven by peptides with varying affinities; however, by increasing the concentration of the peptide, even low-affinity ligands can positively select.177 It has been argued that the recognition of self-peptides in PS must be relatively degenerate. In other words, a single pMHC can trigger the PS of multiple thymocytes;178 therefore, the peptide recognition during PS is cross-reactive.179 This apparent degeneracy of the peptide binding to TCR is the result of a significant contribution to the binding by the MHC molecule.180
Other parameters of a pMHC complex, such as the conformation181 and the concentration182 of the peptide, contribute to the repertoire selection. Despite the evidence that each pMHC complex selects more than one TCR, the large diversity of the fully developed T-cell repertoire depends on a large number of pMHCs183 as well as on the quantity and quality of the stromal cells.184 Peptide diversity during the selection seems to have a greater effect on negative than on PS. In thymus organ cultures, testing with a diverse array of peptides, the addition of 1% of dendritic cells reduced the number of CD4+ T-cells selected by 80% compared to that of the controls in the absence of dendritic cells. Thus, the quantity and quality of the selecting stromal cells have a significant impact on the selected repertoire by multiple peptides.184 At the end of the selection process, mature T lymphocytes exit the thymus and home to the secondary lymphoid organs (see Chapter 11). There are two types of TCR-αβ T-cells, the CD4+ (T helper cell) and the CD8+ (cytotoxic T-cell) and one TCR-γδ cell.
THE α/β T-CELL RECEPTOR COMPLEX
THE MAJOR HISTOCOMPATIBILITY COMPLEX-PEPTIDE COMPLEX
The αβ receptor is formed by two chains, α and β (Fig. 13.5), each consisting of a constant domain, C-α and C-β, and a variable domain, V-α and V-β. The V-α domain is encoded by two gene segments, V-α and J-α, and is homologous to the V domain of the Ig heavy chain (IGH@). The V-β domain is formed by three polypeptides, V-β, D, and J-β. The constant domains correspond to the Ig C domains, but there are certain differences. The C-α-C-β interface is highly polar, whereas that of CL-CH1 is hydrophobic. C-β has a large loop, which extends out to the side of the domain. It has been proposed that it may interact with the coreceptors. The C-α domain has several structural deviations from the C-type Ig domain.
The V domains are very similar to the V domains of an antibody molecule. They contribute to the formation of the TCR-combining site, which is made up of hypervariable loops or complementarity-determining regions CDR 1, 2, and 3 from the α– and β-chains and another loop termed HV-4, which exhibits some hypervariability. The CDR1 and CDR2 are formed by the V segments, which are less polymorphic in the TCR than in IGH@ because fewer V segments are available for TCR, whereas the CDR3 is polymorphic as
a result of the larger number of J segments available for β-chain contributing to CDR3.
a result of the larger number of J segments available for β-chain contributing to CDR3.
 FIGURE 13.5. T-cell receptor complex. The T-cell receptor complex consists of two components: ligand binding (antigen recognition) and signal transduction. The antigen recognition component consists of two polypeptide chains, α and β. Because of their short intracytoplasmic tails, they cannot link themselves to the signal transduction cascade. The signal transduction component consists of the CD3 proteins γ, δ, and ε, members of the immunoglobulin superfamily, and two other proteins forming either a homodimer (two ζ proteins) or a heterodimer (ζ–ε). The γ-, δ-, and ε-chains have one immunoreceptor tyrosine-based activation motif each, and the ζ-chain has three. See text for details. |
The loops of V-β that form the expected antigen-binding site of the TCR are similarly placed as in the IGH@ V region. The chains are linked by a disulfide bond, and the heterodimer is anchored to the cell membrane by the transmembrane region, ending in the cytoplasm by a short (three- to five-amino acid) cytoplasmic tail. Crystallization of the TCR shows that it resembles the Fab fragment of the Ig molecule.187 However, the TCR-αβ is extensively glycosylated with up to seven N-linked sites distributed between the α– and β-chains. The combining site is usually flat, similar to antiprotein antibodies and consistent with the TCR’s function of binding to the generally flat, undulating surface of the pMHC.188 The diversity of the CDR3 is much higher, implying that the function of CD-3 is in peptide discrimination, whereas CDR1 and 2 interact with more conserved structural elements of the MHC. The TCR contains many more J segments and thus are able to increase V-Jα and V-D-Jβ junctional diversity in the CDR3.189 The residues of the TCR that contact the pMHC are always in the apices of the CDR (i.e., for CDR1α, residues 27 to 30; for CDR2α, residues 50 to 52; for CDR1β residues 27 to 30; and for CDR2β residues 52 and 53).
The TCR interacts with peptides bound to MHC molecules (Fig. 13.6). The aminoterminal domains, α1 and α2 of the MHC class I heavy chain, form the binding site for the peptide. The site consists of a floor of eight strands of antiparallel β-pleated sheets, which support two α helices, one contributed by the α1 domain and the other by the α2 domain aligned in an antiparallel orientation. The floor of the groove is supported by two Ig domains from below; one is the α3 domain of the heavy chain and the other the β2-microglobulin. This arrangement forms a groove, on the floor of which lies the peptide from an antigen to be presented to the TCR. Some of the residues of the peptide are exposed above the groove and interact directly with the TCR, whereas others point to the floor of the groove. Depressions in the floor of the groove, known as pockets A through F, interact with some side chains of the bound peptide.190
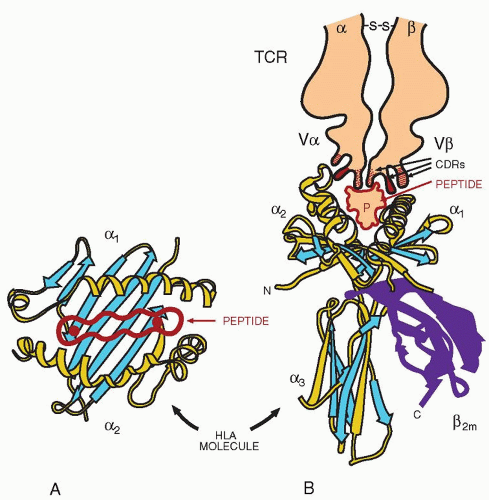 FIGURE 13.6. T-cell receptor (TCR) interaction with HLA/peptide complex. Crystallographic studies of the HLA class I molecule have shown that the two variable domains (α1 and α2) form a groove that binds the peptide that is released as a result of antigen processing. The peptide groove is formed by the helices of the α1 and α2 domains (A). Binding of the peptide is mediated through anchor amino acids near both ends of the peptide (in A). The TCR binds to both the major histocompatibility complex molecule and the peptide. The complementarity-determining region (CDR) 3 of both the α– and β-chains interacts with amino acids of the peptide, whereas the CDR1 and CDR2 interact with the major histocompatibility complex molecule (B). |
For HLA-B27, the side chain of the C-terminal of the bound peptide sits deep in the F pocket, whereas the N-terminal forms strong hydrogen bonds with the hydroxyl groups of conserved amino acids at the end of the groove.191 (For details of interaction with the pMHC, see Chapter 14.) For crystallization of TCR-pMHC, the peptide was attached covalently through a linker to the amino terminus of the β-chain of an MHC class II molecule192 or covalently connected to the amino terminus of the β-chain of the TCR193 and thus tethered to the MHC groove. Such approaches provided stable complexes of HLA-DR1 and HLA-DR4 that permitted their crystallization. Several TCR-pMHCs have been analyzed to clarify the contributions of Vα for the buried surface area (i.e., the interface between TCR and the pMHC). The contribution of Vα is on the average 57% and that of the Vβ 43%.194 The orientation of the TCR over the pMHC is diagonal rather than orthogonal.195 It is more likely that the TCR turns around the MHC molecule by approximately 35° and varies also in its roll in a range of 19° (range 30°).196 As shown by x-ray crystallography, with 2.5 angstrom resolution, of a complex of human αβ-TCR bound to a complex of HLA-A2/HTLV-1 Tax peptide, (HTLV = human T-cell leukemia virus), the initiation of T-cell signaling is favored by a diagonal binding position of the TCR with the supramolecular assembly. This form of interaction of the TCR with its target is in striking contrast to antibody molecules, which use different binding positions even with the same antigen. The diagonal approach of the TCR, to the MHC/peptide complex, evolved to facilitate the initiation of T-cell signaling, inasmuch as the same MHC-bound antigen can be recognized by different TCRs. Unlike antibody molecules, which bind even to the same antigen by many different binding geometries, the TCRs interact with the MHC/peptide complex with a diagonal binding mode. The diagonal binding
geometry has implications for the mechanism of initiation of T-cell signaling, because it places the TCR in a orientation that facilitates the positioning of the CDR1 and CDR2 loops over the α1/α2 helices of MHC class I, and the α1/β1 helices for class II molecules. The Vα domain is critical in setting up these orientations facilitating the read-out of the peptide sequence. Thus the CDR1 and CDR2 interactions of Vα are conservative186 and provide the basic affinity of the TCR. The CDR3 loop is primarily positioned for contact of the peptide in the peptide-binding groove whereas the Vβ interaction is more variable in the C-terminal half of the peptide. The contribution of the individual CDR loops to the interaction varies. CDR1β and CDR2β actually contribute little or nothing to the interaction whereas the CDR3 loops are centrally located and usually dominate the interactions.194 The contribution of the peptide to the interactions with the TCR varies and usually, two to five side chains of the peptide make direct contact with the TCR. These contact points, known as hot spots, are peptide residues that bulge out of the groove which is more prominent and sometimes profound as in MHC class I.197 In the interactions of MHC class II with the TCR, the contributions of the peptide side chains are more uniformly dispersed and the peptides are slightly deeper in the MHC binding groove.197 No single contact dominates the TCR-pMHC interactions, as is often observed in the antigen-antibody interactions. A small number of amino acids dominate the energy landscape in antigen-antibody interaction, as shown by somatic mutations that result in higher-affinity binding.198 Based on these considerations, it appears likely that the CDR1 and CDR2 loops interacting with the helices of the MHC are responsible for PS, and the CDR3 loops probably play a more important role in NS. As an extension of these considerations in delineating the roles of V-region loops of the TCR, the CDR1 and CDR2 loops provide the basic affinity in the interaction, whereas the CDR3 provides the specificity.194 The residues of the peptide that protrude highest from the groove provide the basis for discrimination of peptide and for altering affinity or half-life of the TCR-pMHC interaction.
geometry has implications for the mechanism of initiation of T-cell signaling, because it places the TCR in a orientation that facilitates the positioning of the CDR1 and CDR2 loops over the α1/α2 helices of MHC class I, and the α1/β1 helices for class II molecules. The Vα domain is critical in setting up these orientations facilitating the read-out of the peptide sequence. Thus the CDR1 and CDR2 interactions of Vα are conservative186 and provide the basic affinity of the TCR. The CDR3 loop is primarily positioned for contact of the peptide in the peptide-binding groove whereas the Vβ interaction is more variable in the C-terminal half of the peptide. The contribution of the individual CDR loops to the interaction varies. CDR1β and CDR2β actually contribute little or nothing to the interaction whereas the CDR3 loops are centrally located and usually dominate the interactions.194 The contribution of the peptide to the interactions with the TCR varies and usually, two to five side chains of the peptide make direct contact with the TCR. These contact points, known as hot spots, are peptide residues that bulge out of the groove which is more prominent and sometimes profound as in MHC class I.197 In the interactions of MHC class II with the TCR, the contributions of the peptide side chains are more uniformly dispersed and the peptides are slightly deeper in the MHC binding groove.197 No single contact dominates the TCR-pMHC interactions, as is often observed in the antigen-antibody interactions. A small number of amino acids dominate the energy landscape in antigen-antibody interaction, as shown by somatic mutations that result in higher-affinity binding.198 Based on these considerations, it appears likely that the CDR1 and CDR2 loops interacting with the helices of the MHC are responsible for PS, and the CDR3 loops probably play a more important role in NS. As an extension of these considerations in delineating the roles of V-region loops of the TCR, the CDR1 and CDR2 loops provide the basic affinity in the interaction, whereas the CDR3 provides the specificity.194 The residues of the peptide that protrude highest from the groove provide the basis for discrimination of peptide and for altering affinity or half-life of the TCR-pMHC interaction.
An important point that came out of these crystallographic studies is the role of the water that fills the TCR-pMHC interfaces. This water provides additional complementarity by filling the cavities in the interface, and some of these molecules mediate contact between TCR and pMHC.199 Overall, the consistent feature of TCR-pMHC interaction is that the peptide contributes a smaller portion of the binding interface (21% to 34%) and a smaller proportion of contacts (26% to 47%) than the MHC. The central positions of the peptide play the critical role and those define the peptides as agonists, partial agonists, and antagonists.200 The binding of TCR to the pMHC results in T-cell activation and usually there is a broad correlation among affinity, half-life, and the functional outcomes.201 Mutational analysis of the role of the centrally located residues indicates that in some systems, the biologic activity increases,202 yet in others, the peptide is converted from an agonist to an antagonist;203 nevertheless the affinities of the pMHC for TCR change only marginally.
Contacts on certain hot spots are very sensitive in changing the functional read-out but are not based on changes in the affinity of binding or half-life differences in the TCR-pMHC complexes. It has been argued that affinities usually have been measured in isolated TCR-pMHC complexes, and true affinity measurements may require the presence of coreceptors and signaling components and need to be measured with cellular assays.203
Because changes in the TCR-pMHC complementarity interface are important for triggering biologic reactions, the conformational changes that have been observed in the TCR-complex crystal structures have been considered for initiation of signaling, but their role is not clear.204 Conformational changes contribute to an increase in the binding of a number of other peptides (i.e., an expansion of the repertoire).205
PEPTIDE-MAJOR HISTOCOMPATIBILITY COMPLEX INTERACTION: THE CORECEPTORS
The TCR is not alone in its interaction with the pMHC but is associated with coreceptors (i.e., CD4, CD8, and the CD3 chains). The monomorphic CD3 γ-, δ-, ε-, and ζ-chains, together with the αβ heterodimer form the TCR complex. The CD4 or CD8 act as assistants to the TCRs in the helper or cytotoxic function of the cells, respectively. Therefore they have been known as coreceptors.111 In the current model, the CD4 binds to the same MHC II as the TCR of the CD4+ T-cell and similarly the CD8 binds to the same MHC I as the TCR of the CD8+ T-cell. The binding of the coreceptors occurs with another site of the MHC molecule than that involved in the TCR binding. The TCR binds to the pMHC surface at an angle of 45° to 80° relative to the axis of the two α-helices of MHC,206 which excludes the possibility for direct association of the coreceptors with the TCR that binds the same pMHC. One possibility is that the coreceptors could be linked with TCR in the cytoplasm through signaling molecules (i.e., ZAP70 and LCK).207 Another possibility is that the coreceptor associated with a TCR binds to a different pMHC to which a second TCR binds forming a pseudodimer.208
The CD8 acts as a dimer that includes either two α-chains or one α– and one β-chain, whereas CD4 is a monomer with four Ig-like domains. Both CD8 chains consist of an Ig-like V domain and a long mucinlike stalk. The Ig-like domain binds to the α3 domain of MHC,209 forming also some hydrogen bonds with the α2 domain and the β2M-chain, away from the peptide interface. In CD4, only the fourth Ig-like domain binds directly to pMHC. The ternary complex (TCR-pMHC-CD4/CD8) within the T-cell-APC interface of the immunologic synapse (IS) has a V shape210 (Fig. 13.7). Although the CD8α stalk is longer than the CD8β chain, even in its full extension, it reaches only 50 to 60 Å, which is not long enough to traverse the distance of approximately 100 Å to the TCR-pMHC complex. As a result, the TCR-pMHC has to tilt for the CD-8 to reach the MHC. The stalk of the CD8 chains is heavily glycosylated and changes in glycosylation (which occur after T-cell activation) result in a decrease of binding to MHC.211 The coreceptors enhance TCR signaling by strengthening the stability of the TCR-pMHC complex.212 The V-shaped ternary complex accepts that the coreceptors bind to the same pMHC as the TCR,210 whereas the CD-3 components probably lie inside the “open angel” of the V-shaped structure (Fig. 13.7). Such an arrangement makes possible the association of signaling molecules, such as ZAP-70, LCK, and SRC kinases, with CD4 and CD8. With this topologic model, direct interaction of the coreceptors with TCR is not likely as was previously indicated.213
The CD2 binds to CD58 (LFA-3) in humans based on electrostatic complementarity, with the CD2 surface heavily populated by basic residues, whereas the CD58 is acidic. The interactions span approximately 134 Å, very similar to that of TCR-pMHC. Therefore, the CD2/CD58 interaction in the contact zone between T-cell and the APC, would facilitate the scanning of pMHC by TCR and lower the threshold for TCR triggering in vitro and T-cell activation in vivo.214
CD28 and CTLA4 (CD152) are type I membrane proteins consisting of one moderately to heavily glycosylated V Ig-like domain and are expressed as disulfide-linked homodimers. Their counterreceptors B7-1 (CD80) and B7-2 (CD86) consist of two Ig-like domains, a membrane-proximal C-2 type, and a membrane-distal V-type. B7-2 binds C-28 more effectively than CTLA4 and as a result, enhances costimulatory effects when CD28 and CTLA4 are co-expressed. In contrast, B7-1 binds preferentially CTLA4, and its inhibitory effect would not be affected in the presence of CD28. Delayed expression of B7-1 on APCs appears to be timed to enhance the inhibitory function of
CTLA4. The CTLA4 periodic arrays of crystal lattices enhance the avidity of interaction, whereas the CD28/B7-2 interaction does not have this potential. As a result, during an immune response the CD28/B7-2-activating complexes are 10,000-fold less stable than the inhibitory complexes formed later by CTLA4/B7-1.215
CTLA4. The CTLA4 periodic arrays of crystal lattices enhance the avidity of interaction, whereas the CD28/B7-2 interaction does not have this potential. As a result, during an immune response the CD28/B7-2-activating complexes are 10,000-fold less stable than the inhibitory complexes formed later by CTLA4/B7-1.215
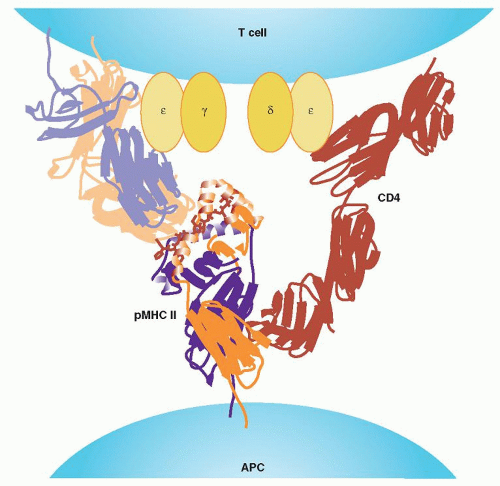 FIGURE 13.7. T-cell receptor (TCR) major histocompatibility complex (MHC) coreceptor interaction. The CD4 and CD8 are coreceptors of the TCR (i.e., “assistants” in the interaction of TCR with peptide-MHC complex [pMHC]). In this function, the N-terminal domain of CD4 (D1) makes contact with the α2 and β2 domains of MHC II. In the CD8, the CD8α subunit of the CD8αβ heterodimer contributes the binding energy, interacting with both the α2 and α3 domains of pMHC I, and CD8β binds only to α3. Therefore, both coreceptors recognize different sites of MHC from the TCR. The length of each receptor is not long enough to reach the MHC binding site, traversing alongside the TCR-MHC complex. It is proposed that TCR-MHC has to tilt for the coreceptor to reach the MHC, forming a V-shaped ternary complex. The CD3 components of the TCR (γ, δ, ε) probably are located inside the V-shaped structure. APC, T-cell-antigen presenting cell. (Courtesy of Dr. G. F. Gao.) |
THE T-CELL RECEPTOR: SIGNAL TRANSDUCTION COMPONENT
The TCR-αβ is accompanied by five other polypeptide chains, collectively known as CD3 proteins: γ, δ, ε, ζ, and ε. They form disulfide-linked heterodimers, such as γδ, εδ, ζε, or a homodimer ζζ216 (Fig. 13.5). The γ, δ, and ε proteins show a significant degree of similarity to one another and consist of an Ig-like domain similar to a C domain, with an intrachain disulfide bond. The extracellular region of the ζ-chain is only nine amino acids long and contains the only cysteine of the molecule, which forms the disulfide bond with another ζ chain or with an ε-chain. In the transmembrane region, the γ, δ, and ε proteins have a negatively charged amino acid complementary to a positively charged amino acid of the transmembrane region of the TCR-αβ chains. The cytoplasmic regions of γ-, δ-, and ε-chains are long, ranging from 40 to 80 amino acids, whereas that of the ζ-chain is longer with 113 amino acids. The ε-chain is a splice variant of the ζ-chain and like a heterodimer with the ζ-chain, exists only in a small number of T-cells. The CD3 proteins have a dual mission in the function of the TCR: escort the receptor to the cell membrane and mediate the signals generated by the TCR-pMHC complex. The complex is assembled in the endoplasmic reticulum, transported to the Golgi apparatus, and then transferred to the plasma membrane. The numbers of the ζ-chain are the rate limiting of the synthesis of the other chains because, although it is synthesized at only 10% of the level of the other chains, it regulates the degradation of a vast majority of the newly synthesized α, β, or CD3 components within 4 hours of their synthesis. The remaining nondegraded chains are long lived and form the complete TCR-CD3 complexes with a limiting role by the ζ-chain. The TCR-CD3 complex lacking the ζ-chain, migrates through the endoplasmic reticulum and Golgi apparatus intact and it is then transported to the lysosomes, where it is degraded. A lysosome-targeting motif has been identified in the δ– and γ-chains and consists of a dileucine-based motif (DKQTLL) and tyrosine-based motif217 in the carboxy terminal region. In the completed complex the TCR α-chain pairs with CD3 δ– and ε-chains, and the TCR β-chain pairs with CD3 γ and ε. The ζ-chain joins the TCR and the CD3 chains in the last stage of the assembly. The topology of the TCR-CD3 complex is shown in Figure 13.5. Two TCR αβ heterodimers are associated with one each of γε and δε heterodimers and one ζζ homodimer. The signal transduction function of the CD3 proteins is based on the presence of one ITAM in each of the γ, δ, and ε proteins and three in the ζ-chain. An ITAM consists of two YXXY/L sequences separated by six to eight amino acids and in the one-letter code for amino acids: Y = tyrosine, X = any amino acid, and L = leucine).218
Phosphorylation of the tyrosines turns the ITAM motifs into docking sites for protein tyrosine kinases, which bind to the ITAM through their SH2 domain (see below). An important protein tyrosine kinase in T-cell activation is the ZAP70, which is recruited to the ITAM motifs of the ζ-chain. The multiplicity of ITAM motifs in the cytoplasmic tails of the CD3 proteins results in signal amplification and increases the sensitivity of the TCR to ligand stimulation. Triplication of a single ITAM motif significantly enhances Ca2+ mobilization, association with ZAP70 and transcriptional activity in the NFAT complex involved in IL2 gene regulation.219 Cross-linking of a single isolated ITAM results in approximately threefold induction in NFAT-regulated activity and cross-linking of a triplicated motif results in an approximately eightfold increase in NFAT-regulated activity (i.e., comparable to intact ζ-chain).
CORECEPTORS TO T-CELL RECEPTOR: CD4 AND C-8 CD4
CD4 is the characteristic marker of αβ T-cells with helper activity and cytokine secretion. The molecule consists of four Ig-like domains, (D-1 to D-4), with domains D-1 and D-3 similar to a V domain and D-2 and D-4 similar to C domains (C-2 type), but with patches of sequences similar to the V domain. In D-1 and D-3 domains, the nine β strands form two β sheets, linked by a disulfide bond. The domains are linearly arranged, forming a rod with limited flexibility.220 The binding of CD4 to MHC II molecules and to HIVgp120-envelop protein was studied by mutational analysis. The binding is mediated by a sequence within the D1 domain of two β strands, C′C″, that form a ridge (residues 35 to 46).221 Phe-43 within this sequence is critical for binding to gp120, as well as for CD4 coreceptor function222 because it provides the major binding energy, whereas the surrounding charged amino acids facilitate specificity.223 Crystal structure of the D1/D2 domains from CD4 complexed with class II molecule shows that the CD4 N-terminal V domain is directed and reaching into the two membrane-proximal domains of the MHC II molecule.224 Both TCR and CD4 are tilted rather than vertically oriented, forming a V-shaped CD4-pMHCII-TCR ternary complex (Fig. 13.7).
In this complex, the antigen-binding groove of pMHC II is no longer parallel to the cell surface but makes an approximately
45° angle with the membrane. However, despite the V shape of the ternary complex, the membrane-proximal domains of each of the components are all roughly vertical, including domain 4 of CD4 and the α2 and β2 domains of pMHC II as well as the C-β domain of TCR. Only the C-α domain of TCR hangs almost parallel above the membrane with its lengthy stalk bridging the space.
45° angle with the membrane. However, despite the V shape of the ternary complex, the membrane-proximal domains of each of the components are all roughly vertical, including domain 4 of CD4 and the α2 and β2 domains of pMHC II as well as the C-β domain of TCR. Only the C-α domain of TCR hangs almost parallel above the membrane with its lengthy stalk bridging the space.
The extracellular fragment of CD4 crystallizes as a dimer associated via the D-4 domains.225 The CD4 dimers are associated with superdimers or dimers of dimers of the MHC II molecules227 and each CD4 molecule of the CD4 dimer interacts with one dimer of MHC II. X-ray crystallography of several MHC class II molecules revealed a structure described as a dimer of heterodimers, or a superdimer. This discovery led to the hypothesis that the MHC class II molecule may interact with the TCR and CD4 as an α/β-2 superdimer, potentially providing more stable and stimulatory interactions than can be provided by the simple αβ heterodimer alone and indeed oligomers of CD4 have been extracted from isolated lymphocytes.226 Amino acid substitutions in the faces of the MHC II molecules that participate in the formation of the superdimers block activation of CD4+ T-cells, implying that superdimer formation is a prerequisite for T-cell activation.227 Stable binding of the T-cell to MHC II requires oligomerization of CD4, facilitated by the D3/D4 domains. Existing evidence suggests that CD4, once it binds to one MHC II molecule, forms tetramers or even larger oligomers and cross-linked lattices228 a process influenced not only by MHC II but also by the TCR.
When CD4 and TCR co-localize for interaction with the same pMHC II molecule, the CD4 brings p56lck, associated with its short cytoplasmic tail to the site of immune recognition. This may constitute one of the main contributions of CD4 to TCR signaling.229
CD8
The CD8 consists of an Ig-like ectodomain, a membrane-proximal stalk (or hinge) region, a transmembrane region, and a cytoplasmic region. The Ig-like domain is involved in the binding to MHC, as shown by crystallographic evidence230 and mutational analysis.231 The CDR loops of the A and B strands of the CD8αα molecule contact the α2, α3, and β2-M domains of MHC I.232 The β-subunit of the CD8 interacts only with the α3 domain, whereas the α-subunit interacts with the α2 and α3 domains.233 In these interactions, the α3 domain is shifted for better accommodation of the CD8 binding.209 Multiple contacts between the coreceptor and MHC I promote the functional contributions of the coreceptor to T-cell activation.195 CD8 enhances cytotoxic T lymphocyte (CTL) activation by promoting the stability of interaction between the APC and the T-cell.234
The α-chain of the CD8 is associated with the p56lck, which is brought closer to the ZAP70 kinase associated with the TCR, a role that has also been assigned to the CD4 coreceptor. The contributions therefore of both coreceptors in ligand recognition and enhancement of TCR signaling are multiple and involve facilitation of TCR clustering, stabilization of the TCR-pMHC complex, and promotion of signaling by bringing together signaling molecules attached separately on the cytoplasmic tails of the TCR and coreceptors.235 The availability of pMHC tetramers has encouraged studies on the function of CD4 and CD8 coreceptors in TCR binding to pMHC. The CD4 is critical for signal transduction with pMHC tetramers,236,237 and 238 but the CD8 coreceptor also contributes to the initial phase of interaction (binding), the duration of interaction (stability), and the delivery of signal transduction.239,240 The final result of CD8 function depends on an epitope of the CD8 involved in the interactions, which are more pronounced with low-affinity ligands. Blockade of CD8-β may affect TCR-C-8 rather than CD8-MHC interaction. The CD8β is more efficient than CD8-α for association with the TCR.241
An important function of the coreceptors in TCR-mediated activation is the demonstration that CD4 and CD8 associate with LAT (linker for activation of T-cells), a 36- to 38-kD membrane-associated adaptor protein, that plays a central role in TCR signaling.242,243 As a result of LAT association with surface coreceptors and coengagement of the TCR with the coreceptors, LAT is phosphorylated and recruits downstream signaling molecules.244 In conclusion, the contribution of the coreceptors in TCR signaling results from (a) the physical approximation of the LCK, which is associated with the coreceptors; (b) phosphorylation of the ITAMs of the CD3 chains of TCR; (c) recruitment of ZAP70; and (d) phosphorylation of LAT, also associated with the coreceptors. Individual TCR molecules are probably associated with either LCK or with LAT.244
T-CELL RECEPTOR AND T-CELL ACTIVATION
Topology of Immune Recognition
The αβ-TCR recognizes short peptides (i.e., eight to ten residues long) for class I-restricted TCRs or 13 to 25 amino acids for class II restricted TCRs. However, approximately only nine amino acids contact the TCR. The peptide is held by the MHC molecule embedded in a groove formed by the α1–α2 domains of MHC I or by the α1–β1 domains of the MHC II (see Chapter 14). Interaction of TCRs initiates signal transduction leading to transcription of genes encoding cytokines in CD4+ T-cells or assembling and mobilizing the lytic machinery in CD8+ T-cells for killing cells infected by viruses, transformed to a malignant state, or being “strangers,” such as in transplanted tissues. The crystal structure of αβ-TCR has been solved and, for several of them in complex with their cognate pMHC ligand.193,195,196,245,246,247 The data support the docking model of the TCR ligand interaction, in which the TCR approaches the peptide held on an MHC platform. In this model, the Vα domain of the TCR is closest to the N-terminal residues of the peptide, whereas the Vβ domain of the TCR is closest to the C-terminal end of the peptide. In this orientation, the TCR Vβ domain contacts the MHC I α1 domain, whereas the TCR Vα domain interacts with the α2 of MHC I. The TCR orientation relative to the long axis of the MHC platform varies between 45° and 80° (i.e., the angle formed between a line passing through the centers of the Vα and Vβ domains of the TCR and a second line defined by the peptide on the MHC platform).248 The peptides in all MHC I structures have their N- and C-termini anchored into two fixed pockets approximately 20 Å apart in the MHC I platform. Longer peptides usually bulge in the center. In MHC II, the peptides assume an extended conformation, and the middle portion is smooth and concaves away from the TCR. Residues 1 and 5 of the peptide (p-1, p-5) in MHC II point toward the TCR and are critical in TCR recognition. The coreceptors bind to the same pMHC ligand as the TCR, but the binding of the coreceptor is independent of the TCR binding and is severalfold weaker.229,249
The TCR, the pMHC, and the coreceptor form a ternary complex that assumes a V shape with the pMHC II at the apex and the TCR and CD4 as the two arms.
Overall, the data from crystallized complexes of TCR with pMHC suggest that TCR docking on the MHC platform involves residues that are conserved within the CDR1 and CDR2 of the TCR and provide a basal level of stabilizing energy in the formation of the complex.250 Variations within the CDR3α and CDR3β loops affect the way the TCRs footprint on the pMHC surface.
Lipid Rafts and the Immunologic Synapse
A model for T-cell activation proposes that a naive or resting T-cell needs to cross several stages to achieve full activation.251 During the first stage, which lasts a few seconds, contacts need to be established by adhesion molecules that overcome charge repulsions between cells. In the next stage, active cytoskeletal
rearrangements bring together accessory molecules and the TCR and concomitantly exclude unligated molecules, such as CD45 (a protein tyrosine phosphatase) as well as CD43. CD43 is a highly glycosylated protein with strong repulsive forces forming a thick glycocalyx “cloud” approximately 45 nm thick around the cells. The distance spanned between the TCR and the MHC-Ag complex is a mere 15 nm; in order for the TCR to sample the complex, it is mandatory that the area of contact between TCR and MHC is clear from interfering tall molecules, such as CD43 and CD45. This process takes minutes. On the other hand, the fundamental signaling units consisting of several molecules, are “loaded” on a lipid raft that is sustained by the cytoskeleton, a process that takes hours. Lipid rafts in evolution are conserved structures that gather receptors involved in signaling in various cell types.252,253,254 and 255 The lipid rafts are cholesterol-dependent microdomains, resistant to solubilization in nonionic detergents at low temperatures.256 The lipids contained within the cellular plasma membrane include glycerophospholipids, glycosphingolipids, and sterols. Rafts consist of sphingolipids and cholesterol, which can move through the more liquid-disordered phase of the membrane containing glycerophospholipids.257 Because of their consistency, lipid rafts have also been referred to as glycosphingo-lipid-enriched membrane microdomains or GEMs. Lipid rafts are anchored by filamentous actin and actin polymerization causes their coalescence.
rearrangements bring together accessory molecules and the TCR and concomitantly exclude unligated molecules, such as CD45 (a protein tyrosine phosphatase) as well as CD43. CD43 is a highly glycosylated protein with strong repulsive forces forming a thick glycocalyx “cloud” approximately 45 nm thick around the cells. The distance spanned between the TCR and the MHC-Ag complex is a mere 15 nm; in order for the TCR to sample the complex, it is mandatory that the area of contact between TCR and MHC is clear from interfering tall molecules, such as CD43 and CD45. This process takes minutes. On the other hand, the fundamental signaling units consisting of several molecules, are “loaded” on a lipid raft that is sustained by the cytoskeleton, a process that takes hours. Lipid rafts in evolution are conserved structures that gather receptors involved in signaling in various cell types.252,253,254 and 255 The lipid rafts are cholesterol-dependent microdomains, resistant to solubilization in nonionic detergents at low temperatures.256 The lipids contained within the cellular plasma membrane include glycerophospholipids, glycosphingolipids, and sterols. Rafts consist of sphingolipids and cholesterol, which can move through the more liquid-disordered phase of the membrane containing glycerophospholipids.257 Because of their consistency, lipid rafts have also been referred to as glycosphingo-lipid-enriched membrane microdomains or GEMs. Lipid rafts are anchored by filamentous actin and actin polymerization causes their coalescence.
Phosphatidylinositol-4,5 biphosphate (PIP2) activates proteins involved in actin-membrane interactions, such as binding of the ezrin-radixin-moesin family to CD44, a protein broadly expressed on detergent-resistant membranes. Detergent-resistant membranes are presumed to be isolated rafts. PIP2 activates actin nucleation and polymerization of raft domains. Raft size is poorly understood because they cannot be visualized by light microscopy without clustering.258 In resting T-cells, the TCR and GM1 are homogeneously distributed. Cross-linking, however, produces large enough aggregates to be visualized using epifluorescence microscopy. Fluorescence resonance energy transfer is sensitive to distances on the order of a few nanometers. Because fluorescence resonance energy transfer was detected between glycosylphosphatidylinositol (GPI)-anchored transferrin receptors interacting with natural ligands, it was concluded that rafts exist but that they are smaller than 70 nm in diameter.258 Others could not find microdomains enriched in GPI-anchored proteins or GM1. Some MHC II molecules loaded with a select set of peptides are detected on microdomains made up of tetraspan proteins, such as CD9, CD63, CD81, or CD82.256 These tetraspan microdomains are recognized by CDw78 antibody (mAb FN1), which originally was believed to recognize an epitope of HLA-class II molecules.
A central feature of lipid rafts is that they allow for the lateral segregation of proteins within the plasma membrane. This provides a mechanism for compartmentalization of signaling components within the membrane, concentrating certain components in lipid rafts and excluding others (Fig. 13.8). Proteins with GPI linkage, such as CD14, CD16, CD48, and CD58, are associated with the outer leaflet of lipid rafts. Cytoplasmic proteins, on the other hand, associate with the inner leaflet of lipid rafts through acylation. Most of the SRC signaling proteins are acylated and raft associated. The vast majority of transmembrane proteins are excluded from rafts constitutively and cannot be induced to partition into rafts on cross-linking.
Some proteins important in T-cell activation, such as LAT, CD4, and CD8, reside on rafts. Other proteins reside constitutively outside the rafts but, when activated, become translocated to rafts. The multichain immune recognition receptor family is an example.259,260 Translocation into rafts after ligand binding appears to be immediate, occurring within seconds of engagement of the receptors, and is selective. For example, CD45, α4 integrin, and IL1 receptor are excluded from rafts and do not translocate into rafts, even on cross-linking.261,262 The transmembrane domains of the receptors appear to have a significant influence on translocation, as exchange or mutation of the transmembrane regions
alters the translocation properties of the receptors. Translocation of the receptor into the lipid rafts does not need actin reorganization. However, stable residency into the rafts depends on interaction with actin cytoskeleton. The constitutive presence of receptors in rafts may be related to their role in cell survival. For example, the pre-TCR is constitutively associated with rafts and, with its signaling, instructs lineage commitment. TCR/CD28 is excluded from rafts in immature thymocytes and signaling leads to apoptosis.253
alters the translocation properties of the receptors. Translocation of the receptor into the lipid rafts does not need actin reorganization. However, stable residency into the rafts depends on interaction with actin cytoskeleton. The constitutive presence of receptors in rafts may be related to their role in cell survival. For example, the pre-TCR is constitutively associated with rafts and, with its signaling, instructs lineage commitment. TCR/CD28 is excluded from rafts in immature thymocytes and signaling leads to apoptosis.253
 FIGURE 13.8. Rafts: elementary units of the immunologic synapse? In the resting state, the multichain immune recognition receptors (MIRR; i.e., T-cell receptor, B-cell receptor, etc.) “float” free on the lipid membrane, or transiently, some may get on board on certain membrane “microdomains,” also known as “lipid rafts.” Interaction with a multivalent ligand aggregates several receptors that accumulate within the rafts, forming multimolecular clusters. On prolonged exposure to the ligand, the MIRR clusters result in raft coalescence with the formation of a large aggregate, the immunologic synapse. Cytoskeletal reorganization from multichain immune recognition receptor and chemokine signaling promotes immunologic synapse formation. (Adapted from a model proposed by Dykstra M, Cherukuri A, Sohn HW et al. Location is everything: lipid rafts and immune cell signaling. Annu Rev Immunol 2003;21:457-481.) |
In addition to their role in signaling, lipid rafts serve as platforms for B-cell receptors (BCRs) to transport bound antigen for processing and presentation. The internalization of BCRs with captured antigen is initiated from rafts.
Retention of receptors in rafts is a mechanism for augmentation of cell activation. The CD19/CD21 complex is excluded from rafts, but its colligation with BCRs by antigen-antibody-complement complexes causes translocation of BCR and CD19/CD21 complex into rafts, where the complex prolongs the retention of BCR within the rafts.263
T-cell activation causes rearrangement of the actin cytoskeleton and polarization of the cell toward the site of activation, such as the APC. This polarization is reflected by re-orientation of the microtubule-organizing center (MTOC) toward the APC.264,265 The TCR is clustered together with associated molecules (CD4, CD8, CD2, CD28, etc.), a process known as capping. Capping depends on actin reorganization regulated by phosphoinositides, which activate Vav (a guanosine diphosphate-guanosine triphosphate [GTP] exchange factor) and the Wiskott-Aldrich syndrome protein (WASP). In patients with Wiskott-Aldrich syndrome, WASP is lacking or markedly reduced, and they have defects in actin polymerization, capping, and antigen-induced proliferative responses.266 WASP regulates the Arp2/3 complex, which mediates actin branching and polymerization.
PIP3 is a strong activator of Vav, which is targeted to detergent-resistant membranes after TCR activation. In Vav-deficient mice, the capping is severely disrupted, and T-cell proliferation is reduced. Raft aggregation is disrupted by the negative regulator of T-cell activation, Cblb (Casitas B-cell lymphoma-b), which is a molecular adaptor and part of the ubiquitin ligation machinery involved in the degradation of phosphorylated proteins. Cblb inhibits TCR clustering and sustained tyrosine phosphorylation. In Cblb-deficient mice, TCR/CD3 stimulation alone can activate receptor clustering without the need for CD28 costimulation.267
Immunologic Synapse
The TCR interactions with pMHC take place in an intercellular junction between the T-cell and the APC. In this junction, signal 1 (TCR) and signal 2 (costimulation) are processed. This interface reveals a dramatic reorganization of signaling components, forming what is called the IS, a term borrowed from Sherrington’s turn-of-the-century definition of the interconnections of neurons as synapses (from the Greek synapsis, meaning joining, linking, connecting).268 The IS relays information across the cell junction in both directions269 organized into two major compartments. The central supramolecular activation cluster (cSMAC), enriched in TCRs and CD28 and the peripheral supramolecular activation cluster (pSMAC), which contains the LFA1 molecule and talin270 (Fig. 13.9). On the side of the APC, correspondingly, the cSMAC contains the pMHC and CD80 (ligand for CD28) and the pSMAC contains the ICAM-1 (counterreceptor for LFA1) and CD58, ligand for CD2. The IS develops over a period of minutes after interactions of the T-cell and the APC. Formation of the synapse depends on an intact cytoskeleton.271 T-cell activation is accompanied by a dynamic reorganization of cortical actin with increase of filamentous actin. These cytoskeletal changes are accompanied by progressive morphologic changes of the T-cells, which first become round, followed by spreading of the cell.272 In the absence of antigen, the T-cell maintains its motility, continues to crawl around the APC and may even leave for another partner. Some receptors on the APC may convey a “danger signal” for the T-cell to pay particular attention and explore the APC in search of antigen.273
Related posts:

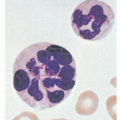

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


