The sideroblastic anemias are a heterogeneous group of disorders
1,2 and 3 uniquely characterized by pathologic iron deposits in erythroblast mitochondria
4,5 (
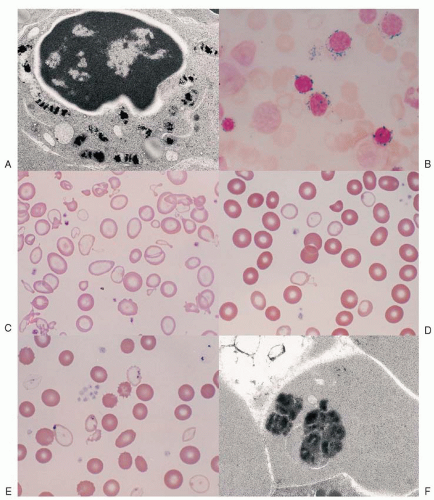
Fig. 24.1A) that are housed within a distinct, mitochondrial ferritin.
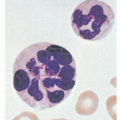
6,7 The iron-glutted mitochondria account for the so-called ring sideroblast, an erythroblast in which numerous Prussian blue-positive granules often appear in a perinuclear distribution, particularly in the later stages of its maturation (
Fig. 24.1B).
The basis for the mitochondrial iron accumulation in the various sideroblastic anemias can be regarded as either insufficient generation of heme as a result of primary defects in the heme biosynthetic pathway or from faults in mitochondrial functions that involve iron pathways, creating an imbalance between mitochondrial iron import and its utilization. Iron delivery to the erythroid cell does not appear to be downregulated in the face of these alterations, and iron continues to be transported normally to mitochondria, where it accumulates.
8,9 Globin synthesis is also reduced, but this effect is secondary, as it can be corrected in vitro by the addition of heme.
10,11Kinetically, the sideroblastic anemias are characterized by ineffective erythropoiesis, like other erythroid disorders with defective cytoplasmic or nuclear maturation.
12,13 Erythroid hyperplasia of the bone marrow is accompanied by a normal or only slightly increased reticulocyte count. The plasma iron turnover rate is increased, but
iron incorporation into circulating red cells is reduced. Red cell survival tends to be normal or slightly reduced. Slight hyperbilirubinemia may be noted, as well as an increase in urobilinogen excretion, as a result of a raised erythropoietic component of the “early-label” bilirubin peak.
14 Thus, it can be inferred that a substantial proportion of the developing ring sideroblasts are nonviable, and their expiration through enhanced apoptotic mechanisms within the marrow
15,16,17 accounts for the kinetic abnormalities.
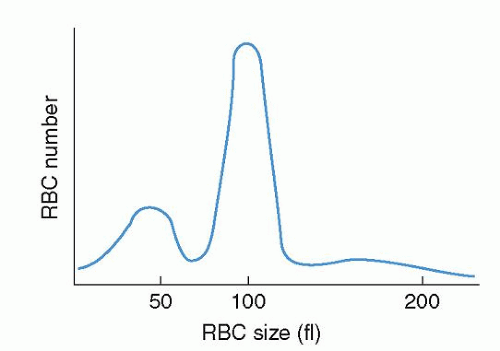
The progeny of surviving ring sideroblasts are often but not always hypochromic and microcytic erythrocytes, a finding that provides morphologic evidence of impaired hemoglobin production as well as an initial clue to the diagnosis. The degree of hypochromia and microcytosis varies considerably from one form of sideroblastic anemia to another (
Fig. 24.1C,D). Often, dimorphism is pronounced, with a hypochromic/microcytic population of cells existing side by side with a normal or even a macrocytic one. The siderotic mitochondria of the developing cell may be retained in some circulating erythrocytes (Pappenheimer bodies) and are regularly found with concurrent hypofunction or absence of the spleen; these cells are the nearly pathognomonic siderocytes in the Wright-stained blood smear
18 (
Fig. 24.1E,F).
A common feature of many sideroblastic anemias that are not reversible is an excess of total body iron. The serum iron concentration is increased, often to the point of complete saturation of transferrin, and the level of serum ferritin roughly reflects the degree of iron overload. The ineffective erythropoiesis mediates increased intestinal absorption of iron by suppressing hepcidin production.
19,20 The consequent iron overload state is called
erythropoietic hemochromatosis, and its clinical and pathologic features and course can rival those of hereditary hemochromatosis
21,22 (
see Chapter 25). The occasional concomitant presence of alleles for genetic hemochromatosis accentuates the iron overload,
23,24 and 25 but their prevalence in patients with sideroblastic anemia does not appear to be greater than in the general population.
22,26,27 and 28Diverse defects affecting the utilization of iron by the developing red cell are reflected in the various forms of sideroblastic anemia (
Table 24.1). Within the congenital group, the majority appear as isolated anemia, the X-linked and a recently identified autosomal recessive type being the most frequent; however, in a large proportion of cases the cause remains unexplained. Very uncommon are several genetically defined syndromic forms involving multiple systems. Acquired sideroblastic anemia is considerably more common than the congenital forms and occurs as a clonal disorder manifesting only anemia or multilineage dysplasia or even myeloproliferative features. Several diverse factors, such as ethanol, certain drugs, copper deficiency, and hypothermia, produce the ring sideroblast abnormality that is fully reversible.
HEME SYNTHESIS IN ERYTHROID CELLS
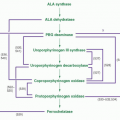
Developing erythroid cells have the greatest requirement of any cell type for heme for hemoglobin, and they produce more than 80% of the heme in the body. The expression and regulation of erythroid heme synthesis are unique in that they are linked (a) to the differentiation events after the signaling action of erythropoietin on erythroid precursors when they acquire the machinery for hemoglobin synthesis, (b) to the availability of iron, and (c) to the production of globin during development of the red cell. As in hepatocytes, 5-aminolevulinate (ALA) synthase and porphobilinogen (PBG) deaminase have considerably lower relative activities than the remaining enzymes in the heme biosynthetic pathway (
see Chapter 6) and are sites of pathway regulation.
62 In contrast to the liver, the relative activity of ferrochelatase, the terminal enzyme in the pathway, appears also to be low in erythroid cells.
62 Furthermore, developing red cells express erythroid-specific isozymes or messenger RNA (mRNA) transcripts of the first four enzymes of heme synthesis, namely ALA synthase, ALA dehydratase, PBG deaminase, and uroporphyrinogen III synthase.
ALA synthase, the first and rate-controlling enzyme of heme synthesis, is generated in the cytosol as a precursor protein with an N-terminal signal sequence that is proteolytically cleaved and processed on transport of the protein into the mitochondrial matrix.
63,64 The mature mitochondrial protein catalyzes the formation of ALA from glycine and succinyl coenzyme A (CoA) and requires pyridoxal 5′-phosphate (PLP) as an obligate cofactor. Recently it was discovered that the erythroid-specific mitochondrial carrier family protein SLC25A38 importing glycine across the mitochondrial inner membrane to likely meet the high molar requirement of this substrate by the ALAS enzyme is
necessary for normal erythroid heme synthesis.
60 Two separate genes encode the ALA synthase isoenzymes.
65,66 The housekeeping gene (
ALAS1), located on chromosome 3,
66,67 is expressed ubiquitously,
68 whereas the erythroid-specific gene (
ALAS2) is on the X chromosome.
65,66,69,70 Expression of the housekeeping gene, at least in hepatocytes, is controlled by glucose levels and is increased by certain steroids, various drugs, and chemicals (
see Chapter 26). This gene is repressed by administration of heme, the end product of the pathway, so that heme levels tightly regulate transcription of its mRNA in a feedback manner.
68 Expression of the erythroid gene is essential for hemoglobin production, and ALAS1 cannot compensate if ALAS2 is lacking.
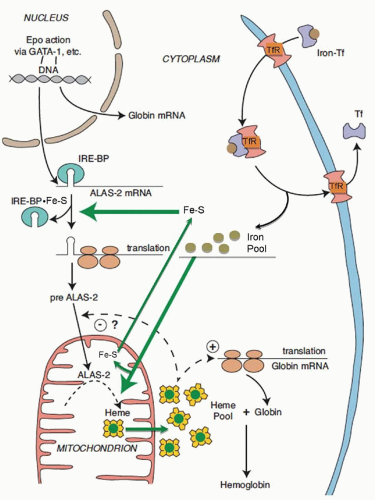
71 ALAS2 is activated and transcribed in concert with other erythroid genes through the action of erythropoietin
63,68 (
Fig. 24.2); it is not repressed by heme but is upregulated by hypoxia.
72 Cellular iron supply may also control ALAS2 mRNA levels,
73 whereas exogenous heme inhibits translation of ALAS2 mRNA,
74 although the mechanisms by which these occur are not known. Translational regulation of erythroid ALA synthase mRNA is mediated through interaction of its
cis-acting iron-responsive element (IRE)
75,76 with iron regulatory proteins (IRP1 and IRP2; also called IRE binding proteins 1 and 2)
77,78,79 that are modulated through iron-sulfur (Fe-S) clusters generated in mitochondria and by cellular iron status
80,81 (
Fig. 24.2) (
see Chapter 23). In this manner, regulation of protoporphyrin production is linked to iron availability and to mitochondrial function for the formation of heme. The low ALA synthase activity observed in erythroid cells in iron deficiency is consistent with such a control mechanism.
82 While the signal sequences of the ALAS1 and ALAS2 precursor proteins contain two heme-binding motifs implicated in regulating translocation of the enzyme into mitochondria by interaction of heme with these motifs
83,84 (
Fig. 24.2), mitochondrial import of ALAS2 does not appear to be affected by heme.
85 Within the mitochondrion, the ALAS2 isoform uniquely associates with the succinyl-CoA synthetase
βA subunit, seemingly to stabilize the ALAS2, to control the generation of its substrate succinyl-CoA, or both.
86 The role of a major splice isoform of ALAS2 that is functional in vivo
87 in the regulation of the enzyme or erythroid heme synthesis remains to be determined.
ALA dehydratase, a cytosolic enzyme, catalyzes the formation of the pyrrole PBG from two molecules of ALA. For this enzyme, two tissue-specific isozymes are produced by a single gene, which contains two promoter regions, generating housekeeping and erythroid-specific transcripts with alternative first noncoding exons (exons 1A and 1B).
88,89 Although both transcripts encode identical polypeptides, the erythroid-regulated form would provide for the production of the large amounts of heme for hemoglobin. Not being a rate-limiting enzyme for heme biosynthesis, its expression in erythroid cells in manyfold excess amounts
62 may also serve as the proteasome inhibitor CF-2 to inhibit protein degradations.
90In the case of PBG deaminase, which acts in the cytosol to form the linear tetrapyrrole hydroxymethylbilane from four molecules of PBG (
see Chapter 6), two tissue-specific isoenzymes also are produced by a single gene.
91 The gene has two overlapping transcription units, each with its own promoter: an upstream ubiquitous promoter and another downstream promoter active only in erythroid cells. Two mRNAs are generated by alternative splicing, one encoding the ubiquitous PBG deaminase isoenzyme and the other encoding the erythroid isoenzyme. To what extent the erythroid-specific enzyme has a regulatory role in the overall production of heme in erythroid cells is not known. In response to erythropoietin or hypoxia, bone marrow PBG deaminase activity increased 3.5-fold, apparently by de novo synthesis.
92The gene for uroporphyrinogen III synthase likewise has two promoters generating housekeeping and erythroid-specific transcripts with unique 5′-untranslated sequences (exons 1 and 2A).
93 As for ALA dehydratase and PBG deaminase, the erythroidpromoter activity is increased during erythroid differentiation. Uroporphyrinogen decarboxylase, the fifth enzyme of the pathway with its site of action in the cytosol, is not known to have erythroid-regulatory features; however, its mRNA is markedly increased in erythroid tissue
94 and the enzyme activity is higher in erythroid cells than in the liver.
62After translation, all three terminal enzymes of heme biosynthesis (coproporphyrinogen oxidase, protoporphyrinogen oxidase, ferrochelatase), like ALA synthase, are transported to their mitochondrial sites of action. Single genes encode these enzymes, and erythroid-specific transcription products are not known for them. However, erythroid-specific regulation of their expression is accommodated by the presence of promoter sequences in their genes for binding of erythroid transcription factors (e.g., GATA-1, NFE-2)
95,96 and 97 to enhance production of these enzymes during erythropoiesis.
98 Ferrochelatase, the last enzyme of the heme synthetic pathway, catalyzes the insertion of iron into protoporphyrin to form heme. A repressor sequence in the promoter region of its gene is believed to be involved in the regulation of the binding of the erythroid transcription factors GATA-1 and NFE-2 to their recognition sites.
97 The activity of ferrochelatase relative to the activity of ALA synthase is in considerable excess,
62,99 but the enzyme becomes rate-limiting as a defective protein in erythropoietic protoporphyria (
see Chapter 26). As it contains an Fe-S cluster, its expression and stability are also dependent on cellular iron levels and intact Fe-S cluster assembly machinery,
100,101 and its activity is regulated by the essential mitochondrial ATPase inhibitory factior 1 (Atpif1) through modulation of mitochondrial pH and redox potential.
102The large amounts of iron required for erythroid heme synthesis are delivered through transferrin receptor-mediated endocytosis of iron transferrin (
see Chapter 23), and iron availability ultimately limits the normal rate of heme synthesis in erythroid cells.
103 High expression of transferrin receptors is also linked to erythropoietin-induced differentiation
68,103; an erythroid-specific isoform of human transferrin receptor has been described
104; and erythroid-active elements have been identified in the promoter of the murine gene.
105,106 During the height of hemoglobinization, the erythroid cell may not depend upon the “standard” mode for regulation of intracellular iron homeostasis via the IRE/IRP system to ensure a maximal supply of iron to mitochondria.
107 With erythroid maturation and the accumulation of cellular hemoglobin, the transferrin receptor number progressively decreases.
103 While surface transferrin receptors and iron uptake are increased in iron-deficient erythroblasts,
108,109 they are not altered in states of impaired heme synthesis, such as in the presence of succinylacetone
110 or in erythroid cells from patients with sideroblastic anemia.
8 Transport of iron out of the endosome by the divalent metal transporter DMT1 requires an endosomal ferrireductase (Steap3) in erythroid cells.
111 How the further transfer of iron to mitochondria and to apoferritin is accomplished is not understood. It may be facilitated by a cytoplasmic chaperone,
112 or through direct interaction of endosomes with mitochondria.
113 Iron is imported across the mitochondrial inner membrane by mitoferrin 1 that is highly expressed in erythroid cells and stabilized by interacting with the ATP-binding cassette protein ABCB10.
114,115 An oligomeric complex of mitoferrin 1, ABCB10 and ferrochelatase appears to facilitate the incorporation of iron into protoporphyrin to form heme.
116 Iron imported into the mitochondrion is also used for the generation of Fe-S clusters, which in part are exported to the cytosol for their addition to IRP1 in the regulation of cellular iron uptake
81 and translation of ALAS2,
117 and for expression of ferrochelatase.
100,101The export of heme out of the mitochondrion into the cytosol for its pairing with globins of hemoglobin and other heme proteins is mediated at least in the mouse by an isoform of the feline leukemia virus C receptor (FLVCR1), named FLVCR1b
118. This heme exporter is essential for erythropoiesis as its loss leads to heme accumulation in the mitochondrion and termination of erythroid differentiation.
The events coordinating the production of globin chains with the rate of heme synthesis are well understood, and appear to occur at more than one level. Heme is required for initiation of globin mRNA translation and it acts by inhibiting a protein kinase called heme-regulated inhibitor (HRI), which inactivates the translational initiation factor 2 (eIF-2
α) in the absence of heme
119 (
Fig. 24.2). Moreover, absence of this kinase adversely modifies the phenotype of disorders of heme synthesis (iron deficiency, protoporphyria) and of globin production (
β-thalassemia).
119 In addition, heme controls globin gene expression at the transcriptional level.
120,121