Target antigens
Antibody medicines
Tumor types
Mechanisms of tumor killing
Hematopoietic cell differentiation antigens
CD20
Rituximab
Non-Hodgkin’s lymphoma
ADCC
Tositumomab
Non-Hodgkin’s lymphoma
131I
Ibritumomab
90Y
CD22
Inotuzumab
Non-Hodgkin’s lymphoma
Ozogamicin
CD30
Brentuximab
Hodgkin’s lymphoma
Monomethyl auristatin E
CD33
Gemtuzumab
Acute myelogenous leukemia
Ozogamicin
CD52
Alemtuzumab
Chronic lymphocytic leukemia
CDC
Chemokine receptor
CCR4
Mogamulizumab
Adult T cell leukemia/lymphoma
ADCC
Tyrosine kinase receptors
EGFR
Cetuximab, panitumumab, nimotuzumab, and 806
Glioma, lung, breast, colon, and head and neck tumors
Inhibition of growth signaling, ADCC and CDC
ERBB2 (HER2)
Trastuzumab and pertuzumab
Breast, colon, lung, ovarian, and prostate cancers
ADCC
ERBB3
MM-121
Breast, colon, lung, ovarian, and prostate cancers
Inhibition of growth signaling
VEGFR
IM-2C6 and CDP791
Epithelium-derived solid tumors
Inhibition of growth signaling
MET
AMG 102 and METMAB
Breast, ovary and lung cancers
Inhibition of growth signaling
IGF1R
AVE1642, IMC-A12, MK-0646, and R1507
Glioma, lung, breast, head and neck, prostate, and thyroid cancers
Inhibition of growth signaling
EPHA3
IIIA4
Lung, kidney and colon cancers, melanoma and glioma
Disrupting the tumor stroma and microvasculature
Death receptors
TRAILR1 (DR3)
Mapatumumab
Colon, lung and pancreas tumors and hematological malignancies
Death signal
TRAILR2 (DR4)
HGS-ETR2 and CS-1008
RANKL
Denosumab
Prostate cancer and bone metastases
Blockade of RANKL/RANK interaction
Glycoproteins
CEA
Labetuzumab
Breast, colon, and lung cancers
131I
EpCAM
Adecatumumab
Breast and prostate cancers
ADCC and CDC
CA-125
Oregovomab
Ovarian cancer
Enhancement of tumor-specific humoral and cellular immune responses
CAIX
cG250
Renal cell carcinoma
131I, 90Y
Others
VEGF
Bevacizumab
Tumor vasculature
Disrupting the tumor microvasculature
Integrin aVb3
Etaracizumab
Tumor vasculature
Integrin a5b1
Volociximab
Tumor vasculature
FAP
Sibrotuzumab
Colon, breast, lung, pancreas, and head and neck cancers
Disrupting the tumor stromal fibroblasts
In contrast, target antigens recognized by antibodies listed in Table 17.2 are immunomodulatory receptors or ligands (Pardoll 2012; Weber 2010; Ngiow et al. 2011a). These receptors, such as cytotoxic T lymphocyte-associated antigen 4 (CTLA-4; also known as CD152), programmed cell death protein 1 (PD-1; also known as CD274), and lymphocyte activation gene 3 (LAG3), are expressed in activated T cells (Schwartz 1992; Lenschow et al. 1996; Rudd et al. 2009; Ishida et al. 1992), and the ligands, including CD80 (also known as B7-1), CD86 (also known as B7-2), and PD-1 ligand 1 (PD-L1, also known as B7-H1 and CD273), are commonly expressed on antigen-presenting cells (APC), tumors, or non-transformed cells in the tumor microenvironment (Zou and Chen 2008). Over-expression of these molecules and their receptor/ligand pathways are a major cause of exhaustion in CD8 T cells. Exhausted CD8 T cells are functionally deficient and have decreased proliferative capacity, cytokine production, and cytotoxic activity and are metabolically deficient (Barber et al. 2006). The antibodies can block the receptor/ligand pathway between infiltrating T cells in tumors and the tumor cells to restore T cell cytotoxic activity against the tumors. Indeed, significant clinical effects by single drug therapy has been reported for ipilimumab (anti-CTLA-4), nivolumab/BMS‑936558 (anti-PD-1), lambrolizumab/MK-3475 (anti-PD-1), and BMS-936559 (anti-PD-L1) (Hodi et al. 2010; Topalian et al. 2012; Hamid et al. 2013; Brahmer et al. 2012). Thus, turning the immune balance to an active state by blocking immune inhibitory signals with antagonistic antibodies is a new direction for the development of antibody drug therapy against tumors.
Table 17.2
Therapeutic antibodies targeting immune checkpoints
Antibodies | Targets | Counterparts | ||
|---|---|---|---|---|
Antigens | Cells | Antigens | Cells | |
Ipilimumab | CTLA-4 | T cell | CD80 or CD86 (B7-1) (B7-2) | APC |
Tremelimumab | ||||
Nivolumab (BMS-936558) | PD-1 | T-cell | PD-L1 or PD-L2 (B7-H1) (B7-DC) | APC |
Pembrolizumab (MK-3475) | ||||
CT-011 | ||||
AMP-244 | ||||
BMS-936559 | PD-L1 (B7-H1) | Tumor | PD-1, CD80 | T cell |
MEDI4736 | T cell | |||
BMS-986016 | LAG3 | T cell | MHCII | APC |
MGA271 | B7-H3 | APC | ? | T cell ? |
Under development | B7-H4 | APC | ? | T cell ? |
Under development | TIM3 | T cell | Galectin 9 | Tumor |
BMS-986015 | KIR | T cell | MHCI | Tumor |
Regulation of regulatory T cells (Tregs) is also considered important for the development of new therapeutics against tumors because many reports indicate infiltrating Tregs in tumors suppress tumor immune responses and Treg depletion by administration of CD25 mAb causes tumor regression in a mouse tumor model (Onizuka et al. 1999; Ohmura et al. 2008). CCR4 is commonly expressed on Tregs; thus, mogamulizumab can diminish not only ATLL cells but also normal Tregs in vivo (Yano et al. 2007; Ishida and Ueda 2006, 2011; Ishida et al. 2003). Therefore, mogamulizumab might be a unique regulator of immune balance to resolve tumor immune suppression, resulting in new therapeutics for tumors. This review focuses on the regulation of Tregs by mogamulizumab and discusses the theory and possibility of a new tumor therapy using mogamulizumab compared with antibodies to immune checkpoint-related molecules such as ipilimumab, nivolumab, and BMS-936559.
17.2 Regulation of Immune Balance in Immune Checkpoints
Immune checkpoints during the sequential steps of immune responses are crucial events to maintain self-tolerance and to protect tissues from damage caused by hyperimmune reactions during pathogenic inflammation (Pardoll 2012; Zou and Chen 2008). CTLA-4 is transiently expressed on effector T cells after activation and functions as an inhibitory receptor in the first immune checkpoint, which regulates antigen presentation from dendritic cells (DC) in primary or secondary lymph nodes (Zou and Chen 2008). When antigen is presented by DCs to naïve or memory T cells, T cells recognize antigen by binding to the T cell receptor (TCR) and epitope peptide complexed with major histocompatibility complex (MHC) expressed on the DC. Simultaneously, co-stimulatory receptor CD28, exclusively expressed on T cells, binds to CD80/CD86 on DCs and stimulates immune responses (Zou and Chen 2008). CTLA-4 shares identical ligands, CD80/CD86, with CD28, and counteracts the activity of CD28 (Schwartz 1992; Rudd et al. 2009). Although the mechanisms of T cell suppression by CTLA-4 are not clear, two different mechanisms have been demonstrated. CTLA-4 is thought to compete with CD28 in binding CD80 and/or CD86 because the binding affinity of CTLA-4 for both ligands is higher than that of CD28 (Linsley et al. 1994; Egen and Allison 2002). Another explanation is the activation of protein phosphatases, Src homolog domain-containing phosphatase 2 (SHP2, also known as PTPN11) and protein phosphatase 2 (PP2, also known as PP2A), which inhibit kinase signaling downstream of TCR and CD28 (Schwartz 1992). CTLA-4 is constitutively expressed on Tregs as well as CD8+ cytotoxic T cells and CD4+ T helper (Th) cells, and is a target gene of the forkhead transcription factor FOXP3 (Hori et al. 2003; Fontenot et al. 2003), the master gene of Treg. FOXP3 regulates the differentiation and functions of Treg (Hill et al. 2007; Gavin et al. 2007) and is one of the mechanisms of effector T cell suppression that downregulates CD80 and CD86 on APCs. This is caused by the binding of CTLA-4 on Treg counteracting CD28 binding CD80/CD86, resulting in the inhibition of antigen presentation to T cells (Vignali et al. 2008) (Fig. 17.1).
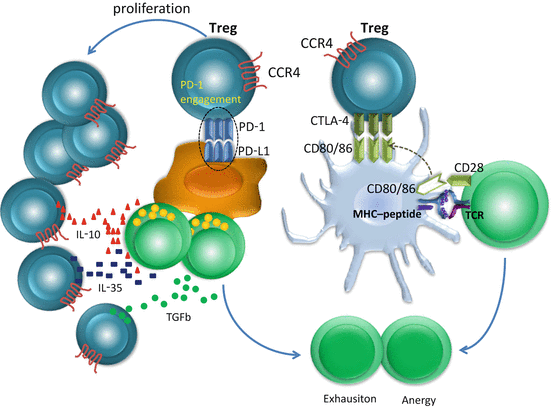
Fig. 17.1
T cell exhaustion mechanisms induced by regulatory T cells (Tregs). Tregs constitutively express cytotoxic T lymphocyte-associated antigen (CTLA)4. The affinity of CTLA-4 to CD80 or CD86 is higher than CD28, thus CTLA-4 outcompetes CD28, and co-stimulatory signals via CD28 are blocked in antigen-stimulated T cells, resulting in anergy. Tregs highly express programmed cell death protein 1 (PD-1), the proliferation of Tregs is enhanced in the presence of ligand, and the effector T cells are exhausted by the release of interleukin (IL)-10, IL-35, and transforming growth factor (TGF)-β from Tregs
PD-1 is another important co-inhibitory receptor stably expressed on effector-phase T cells, which regulates T cell activation by interactions with PD-L1 and/or PD-L2 expressed on tumor cells, APC, and non-transformed cells in the tumor microenvironment (Dong et al. 2002; Konishi et al. 2004; Curiel et al. 2003; Kuang et al. 2009). PD-1 engagement inhibits kinase signaling downstream of TCR through phosphatase SHP2 (Dong et al. 2002). PD-1 is also highly expressed on Tregs and enhances the proliferation of Tregs in the presence of ligand (Freeman et al. 2000; Francisco et al. 2009).
As well as the molecules described above, various inhibitory receptors and ligands are involved in immune checkpoints. T cell membrane protein 3 (TIM3), LAG3 (also known as CD233), B and T Lymphocyte attenuator (BTLA; also known as CD272), adenosine A2a receptor (A2aR), and the family of killer inhibitory receptors are inhibitory receptors expressed on T cells. Galectin 9, MHC class II, herpesvirus entry mediator (HVEM), adenosine, and subsets of human leukocyte antigens (HLA) have been respectively identified as their ligands (Pardoll 2012). These receptors are also expressed on activated T cells, and the ligands are expressed on many types of tumor cells (Ngiow et al. 2011a; Grosso et al. 2007; Cedeno-Laurent and Dimitroff 2012; Fourcade et al. 2012; Baixeras et al. 1992). Furthermore, antibodies to these enhance anti-tumor immunity in mouse models and human in vitro experiments (Baghdadi et al. 2013; Ngiow et al. 2011b). LAG3 is expressed on Tregs as well as effector T cells and enhances Treg functions. A2aR functions in the development of Tregs by inducing FOXP3 expression in CD4+ T cells (Huang et al. 2004; Zarek et al. 2008).
17.3 Clinical Application of the Blockade of Immune Checkpoints
Thus, co-inhibitory receptors/ligands regulate immune responses of effector T cells at sites of inflammation or tumors (Fig. 17.2), and drugs, such as antibodies and small molecules, which block receptor/ligand interactions are expected to be novel beneficial drugs for tumor therapy (Fig. 17.3). Clinical trials of anti-CTLA-4 humanized mAb, ipilimumab, and tremelimumab were conducted from 2000. Although objective clinical responses were observed in ~10 % of patients with melanoma using both antibodies, no survival benefit was observed in comparison with standard melanoma chemotherapy treatment (dacarbazine) in a randomized phase III trial for tremelimumab (Ribas 2010). However, in a randomized, three-arm phase III trial of patients with advanced melanoma using (1) gp100 peptide vaccine alone, (2) gp100 plus ipilimumab, or (3) ipilimumab alone, 3.5-month survival benefit was observed in the ipilimumab alone group compared with the gp100 peptide vaccine alone or gp100 plus ipilimumab groups (Hodi et al. 2010). Furthermore, Grade 3 or 4 immune-related adverse events (AEs) occurred in 10–15 % of patients treated with ipilimumab, including deaths associated with immune-related AEs. Ipilimumab was the first drug to demonstrate a survival benefit for metastatic melanoma, and was approved by the US Food and Drug Administration (FDA) for the treatment of advanced melanoma in 2010. Both humanized mAb to PD-1 (nivolumab/BS936558) and PD-L1 (BS936559) demonstrated very promising results in a phase I clinical trial for multiple tumors (Topalian et al. 2012; Brahmer et al. 2012). Objective response rates to BMS936558 were observed in 18 % of patients with non-small cell lung cancer (14 of 76 patients), 28 % of patients with melanoma (26 of 94 patients), and 27 % of patients with renal cell cancer (9 of 33 patients). Interestingly, no objective response was observed in 17 patients with PD-L1-negative tumors, although 9 of 25 patients (36 %) with PD-L1-positive tumors had an objective response. In a phase I trial using anti-PD-L1 (BMS936559), an objective response (a complete or partial response) was observed in 17 % of patients with melanoma (9 of 52), 12 % of patients with renal cell cancer (2 of 17), 10 % of patients with non-small cell lung cancer (5 of 49), and 6 % of patients with ovarian cancer (1 of 17). Furthermore, responses were durable for both antibodies. Responses lasting 1 year or more were shown in more than half of the patients with 1 year or more of follow-up. Immune-related AEs occurred in 14 % of patients treated with anti-PD-1 and 9 % of patients treated with anti-PD-L1. Interestingly, immune-related AEs frequently occurred in the skin and intestine in patients receiving any of ipilimumab, nivolumab, or BMS936559.
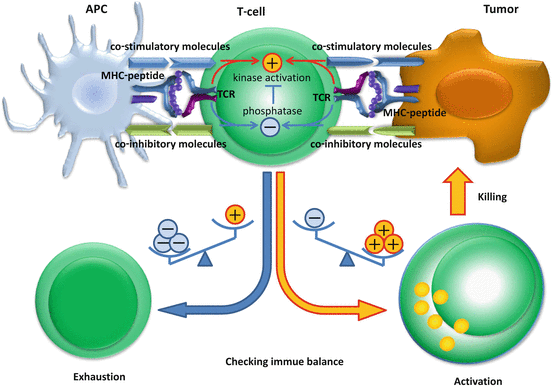
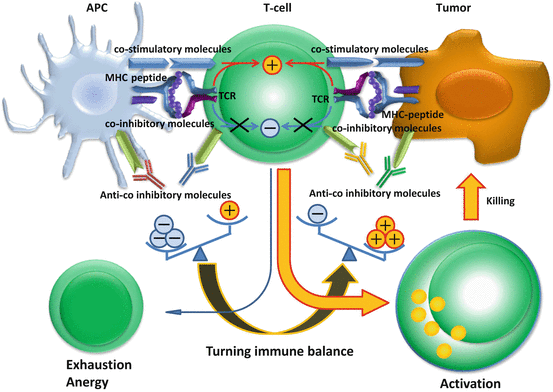
Fig. 17.2
Immune checkpoint determines the balance of stimulatory and inhibitory signals and the future state of T cells. After T cells recognize antigen epitope peptides presented on human leukocyte antigen (HLA) class I molecules, the signal is transduced in addition to co-stimulatory and co-inhibitory molecule signaling. Co-stimulatory receptors (CD28, CD137, ICOS, OX40, CD27) and co-inhibitory receptors [cytotoxic T lymphocyte-associated antigen (CTLA)-4, programmed cell death protein 1 (PD-1), T cell membrane protein 3 (TIM3), killer cell immunoglobulin-like receptor (KIR), lymphocyte activation gene 3 (LAG3), adenosine A2a receptor (A2aR)] are expressed on T cells, and their co-stimulatory ligands (CD80, CD86, CD137L, B7PR1, OX40L, CD70) and co-inhibitory ligands [CD80, CD86, PD-1 ligand (PD-L) 1, PD-L2, galectin 9, major histocompatibility complex (MHC) class II, adenosine) are expressed on antigen-presenting cells or tumor cells. The balance of stimulatory and inhibitory signals determines whether T cells are activated or exhausted. Once tumor-specific T cells are activated, they elicit killing activity to the tumor cells
Fig. 17.3
Antibodies to immune checkpoint molecules block co-inhibitory signals turning the immune balance to an active state. In the tumor site, the immune balance is considered inhibitory, caused by inhibitory signals that exceed stimulatory signals. Antagonistic antibodies such as ipilimumab [anti-cytotoxic T lymphocyte-associated antigen (CTLA)-4], nivolumab [anti-programmed cell death protein 1 (PD-1)], lambrolizumab (anti-PD-1), and BMS936559 [anti-PD-1 ligand 1 (PD-L1)] block co-inhibitory signals of T cells, turning the immune balance towards an active state. Exhausted T cells in the tumor site show restored tumor killing functions and kill tumor cells
17.4 Immune Tolerance in Tumors Caused by Regulatory T Cells
It is clarified that FOXP3 elicits Treg functions (Hori et al. 2003; Fontenot et al. 2003). Because FOXP3 knockout mice develop autoimmune diseases involving multiple organs, and patients with FOXP3 homozygous mutations develop immunodysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome (Fontenot et al. 2003; Bennett et al. 2001), Tregs are considered to be crucial for the maintenance of self-tolerance and for maintaining immune balance to prevent damage of healthy cells from excessive immune reactions in cooperation with the immune checkpoint system described above. It was reported that Tregs are recruited and accumulate in tumor tissues by chemokine-induced chemotaxis via interaction between CCR4 and its ligands produced by cells of the tumor microenvironment (Curiel et al. 2004; Zou 2006). Hodgkin’s lymphoma (HL) is a representative example of Treg recruitment to the tumor site by chemokine-induced chemotaxis resulting in immunosuppression. HL is characterized by the presence of a small number of tumor cells in a rich background of T and B cells, macrophages, and other inflammatory cells (Küppers 2009). The question of why a very small number of HL cells can survive in the presence of a large excess of host immune cells was unclear. HL cells express high levels of the CCR4 ligand and thymus and activation-regulated chemokine (TARC)⁄CCL17 (van den Berg et al. 1999), and elevated serum levels of TARC⁄CCL17 were reported as an unfavorable prognostic factor in patients with HL (Weihrauch et al. 2005). Considering these findings, we have shown that HL tumor cells attracted CD4+CCR4+ T cells by interactions with TARC⁄CCL17 and macrophage-derived chemokine (MDC)⁄CCL22. Migratory CD4+ cells attracted by HL tumor cells were hyporesponsive to TCR stimulation and suppressed the activation and proliferation of effector CD4+ T cells in an autologous setting in vitro. Furthermore, double staining showed that HL cells in the affected lymph nodes were surrounded by a large number of lymphocytes expressing both CCR4 and FOXP3 (Ishida et al. 2006). Collectively, these findings imply that the migratory cells induced by HL function as Tregs to create a favorable environment for the tumor cells to escape from host immunity. It was proposed that Treg-mediated immunosuppression is a crucial tumor immune-evasion mechanism in ovarian, gastric, breast, and pancreatic cancers, and may be one of the main obstacles to successful tumor immunotherapy (Curiel et al. 2004; Zou 2006). Therefore, Tregs are a potential target for novel tumor therapy by inhibition of immunosuppression in the tumor site.
Related posts:
 A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
 PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
 Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
 Development of Personalized Combination Cancer Immunotherapy Based on the Patients’ Immune Status
Development of Personalized Combination Cancer Immunotherapy Based on the Patients’ Immune Status
 Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


