Platelet Function in Hemostasis and Thrombosis
David C. Calverley
PLATELET ADHESION AND ACTIVATION
Primary hemostasis and arterial thrombosis are the results of a complex series of cell-cell, cell-protein, and protein-protein reactions that involve platelets, leukocytes, endothelium, subendothelial matrix, and plasma proteins, such as fibrinogen, von Willebrand factor (vWF), and others. The consequences of arterial thrombosis include such events as myocardial infarction (MI), unstable angina, and stroke. These clinicopathologic entities and their associated cellular physiologic mechanisms that are outlined in this chapter collectively account for the largest cause of morbidity and mortality in the Western world.
Platelet adhesion to exposed subendothelium is a complex multistep process that involves a diverse array of adhesive ligands (vWF, collagen, fibrinogen/fibrin, fibronectin, thrombospondin, laminin, and vitronectin) and surface receptors (gpIb/V/IX, gpVI, integrins αIIbβ3, α2β1, α5β1, and α6β1).1,2,3,4,5,6,7,8,9,10,11,12 and 13 The specific ligand/receptor players in primary platelet adhesion are largely dependent on the arterial flow conditions present.5,14 As such, in larger arteries and veins, platelet adhesion to the vessel wall is thought to involve fibrillar collagen, fibronectin, and laminin. There are at least 25 forms of collagen, and several of these are present in the blood vessel wall, whereas one (type IV) is present in the subendothelial basement membrane.15 In the high-shear conditions present in smaller arteries, platelet tethering is dependent on the unique shear-dependent interaction between gpIb/V/IX and subendothelial vWF. The subsequent rapid platelet deceleration allows for other ligand-receptor interactions such as collagen and α2β1 that have slower binding kinetics and take on the role of mediating firm platelet adhesion. A metalloprotease, ADAMTS13, cleaves vWF; this cleavage prevents the accumulation of ultrahigh-molecular-weight multimers that would otherwise cause spontaneous platelet clumping and arterial thrombosis.
Following initial platelet adhesion, subsequent platelet-platelet interaction (aggregation) is mediated by two receptors, gpIb/V/IX and αIIbβ3, and their respective contributions are dependent on the flow conditions present. In high-shear stress conditions, gpIb/V/IX receptor and vWF ligand action are predominant, with fibrinogen playing a stabilizing role. At low-shear conditions, fibrinogen is thought to be the primary ligand supporting platelet plug formation through its interaction with αIIbβ3. It has been shown that thrombus formation can take place in the absence of vWF and fibrinogen, and this supports the idea that a third ligand directed to the αIIbβ3 receptor may also exist in vivo.16
Platelet Glycoprotein Ib Complex-von Willebrand Factor Interaction and Signaling
It has long been recognized that the interaction of the platelet glycoprotein (gp) Ib “complex” (including the single-chain polypeptides gpIbα, gpIbβ, gpIX, and gpV) with its primary ligand, vWF, is the receptor-ligand pairing that initiates platelet adhesion followed by a cascade of events leading to pathologic thrombosis or physiologic hemostasis. A unique aspect of this receptor-ligand interaction is that it requires the presence of high arterial shear rates to take place, thus explaining the predisposition of plateletrich “white clots” in the arterial circulation over clots found in the venous circulation, with its relatively lower shear forces, in which clot formation takes place independent of the gpIb complex.
Using a parallel-plate flow cytometer, platelet interaction with subendothelial vWF has been characterized as occurring in a biphasic fashion.17 In this respect, the rate of translocation of platelets from blood to the endothelial cell surface increased linearly up to wall shear rates of 1,500 s-1, whereas the translocation rate remained relatively constant with the wall shear rate between 1,500 and 6,000 s-1. This ability to mediate translocation or rolling of the platelet on vWF is contingent on the gpIb complex, and mammalian cells expressing either the full complex or a complex lacking the gpV subunit were able to roll onto vWF in a gpIbα-chain-dependent manner.18,19
It is clear that arterial thrombus formation is contingent on both the presence of high wall shear rates and interaction between the gpIb complex and vWF. Studies involving the endpoint of realtime thrombus formation that involved comparison of blood from both patients with Bernard-Soulier syndrome (which lacks platelet gpIb complex) and severe (type 3) von Willebrand disease versus normal blood led to the conclusion that gpIb and vWF interaction was required for platelet surface interaction at high shear rates (>1,210 s-1), whereas normal thrombus formation at lower shear rates (<340 s-1) was possible with blood deficient in either gpIb or vWF.20 In normal blood, thrombus formation was accelerated as shear rate increased, and this served to verify the unique shear flow dependence of this receptor-ligand interaction.
The gpIb complex consists of four transmembrane subunits, each of which is a member of the leucine-rich repeat protein superfamily that participates in cell-matrix interactions throughout nature. Each of the four subunits contains one or more tandem, 24-amino acid leucine-rich repeats flanked by conserved disulfide loop structures at both the N and C termini of the repeats.21 gpIbα is covalently associated with the gpIbβ chain through disulfide linkage of cysteine residues, and both of these chains are noncovalently associated with gpIX in a 1:1 ratio and with gpV in a 2:1 ratio22,23 and 24 (Fig. 17.1).
vWF is a large multimeric glycoprotein that circulates in plasma and is also found in platelets and the Weibe-Palade bodies of endothelial cells. Mature vWF is a 2,050-amino acid subunit that is disulfide linked into large multimers. It contains three adjacent A domains in the N-proximal half of the peptide that collectively regulate the adhesion of platelets to subendothelial matrix. In this respect, the A1 and A3 domains bind to different matrix collagens, whereas the A1 domain contains the binding site for the gpIb complex.25 The A1 domain is the primary role player in platelet adhesion, because this part of the molecule is believed to change its conformation in response to immobilization and high-shear forces, thus making it a high-affinity ligand for the gpIb complex receptor.26,27 It has also been suggested that shear stresses may induce conformational changes in the gpIb complex that may be important in increasing its affinity for vWF.28 Through the simultaneous binding of collagen and platelets, vWF can serve as a molecular bridge between platelets and the subendothelial matrix mediating platelet adhesion to the vessel wall. Thus by associating with matrix proteins vWF mediates rapid and reversible platelet adhesion that promotes the rolling of these cells along the surface of vascular injury.
To further add to the mystery behind the mechanism of shear dependence in gpIb-vWF interaction, in vitro activation of vWF and binding to the gpIb complex occur with generally very low affinity without shear, whereas this shear-free binding exhibits high affinity in the presence of the vancomycinlike antibiotic ristocetin, and viper venom proteins, such as botrocetin. It is interesting that studies incorporating anti-gpIbα and anti-vWF domain A1 antibodies have suggested that ristocetin and botrocetin each
appear to use different receptor and ligand-binding sites to facilitate the vWF-gpIb complex interaction.29,30 and 31
appear to use different receptor and ligand-binding sites to facilitate the vWF-gpIb complex interaction.29,30 and 31
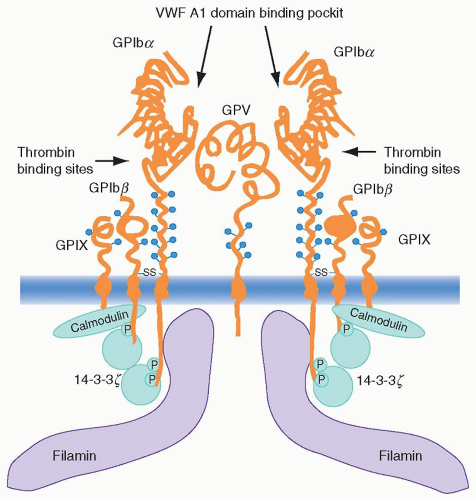 FIGURE 17.1. Schematic illustration of the glycoprotein (GP) Ib/V/IX complex and associated proteins. The N-terminal domain of GPIba contains binding sites for von Willebrand factor (vWF) and thrombin. The cytoplasmic domain of GPIba contains the phosphorylation-dependent 14-3-3 binding sites, and also the binding site for filamin that links GPIb-IX to the actin cytoskeleton underlining the membrane. The cytoplasmic domain of GPIbβ is phosphorylated by protein kinase A. (PKA) and this phosphorylation is required for 14-3-3 binding. The membrane proximal region in the cytoplasmic domain of GPIbβ and GPIX contains calmodulin-binding sites. From Du, X. Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. LWW, Curr Opin Hematol 2007;14:262, Figure 2, with permission. |
The binding interaction between vWF and gpIb appears to involve at least three distinct regions within the N-terminal 282 residues of gpIb. Each of these regions appears to be responsible for either direct binding to vWF or modulating its affinity for the ligand.32 In this respect, one region (His 1 to Glu 282), which includes a cluster between residues Asp 252 and Arg 293 containing sulfated tyrosine residues and important anionic residues, appears to be predominantly responsible for vWF-gpIb complex interaction in the presence of botrocetin over ristocetin.33,34 and 35
The second region contains the disulfide loop between Cys 209 and Cys 248 along with two naturally occurring mutations (Gly 233 to Val and Met 239 to Val) and two additional mutation sites identified in the laboratory (Asp 235 to Val and Lys 237 to Val) that can individually lead to expression of the pseudo- or platelettype von Willebrand disease phenotype.30 This disorder is associated with a gain-of-function gpIbα on platelets that adheres to vWF in the presence of lower concentrations of ristocetin (0.3 to 0.5 mg/ml) than are required for the wild-type phenotype (1.5 g/ml). It is analogous to type 2B von Willebrand disease, in which high-molecular-weight vWF multimers are absent from the plasma, and similar gain-of-function mutations have been localized to the Cys 509 to Cys 695 disulfide loop of vWF exon 28.
The third region includes the N-terminal flanking sequence of the leucine-rich repeat (LRG) motifs and the LRGs themselves. Mutations involving single amino acid residues within these LRGs account for some cases of the congenital bleeding disorder, Bernard-Soulier syndrome, in which the gpIb complex binds poorly or not at all to vWF. Evidence using mammalian Chinese hamster ovary cells expressing loss-of-function proteins and anti-gpIbα monoclonal antibodies has suggested that the more N-terminal LRGs may play a more direct role in interaction with vWF.36
Glycoprotein Ib Complex Interaction with Thrombin
Recent studies have examined the potential role of the gpIb complex in thrombin-mediated platelet activation. The physiologic significance of the interaction of thrombin with the complex has remained relatively controversial. gpIbα contains a well-characterized high-affinity binding site for thrombin, and thrombin is also capable of cleaving gpV near the surface to release a soluble fragment.37
Recent studies have suggested that a relationship exists between thrombin binding to the gpIb complex and cleavage of the seven-transmembrane G-protein-coupled protease-activated thrombin receptor, protease-activated receptor (PAR)-1 (see section “Platelet Thrombin [Protease-Activated] Receptors and Signaling”), and that accelerated coagulation on the surface of a developing thrombus is a downstream consequence of thrombin-gpIb interaction because of enhanced phospholipid exposure.38,39 These studies support a procoagulant role of this thrombin-gpIb pairing and this activity also plays an important role in the generation of platelet microparticles.40,41 In contrast, studies using gpV-null mice have suggested that the gpV subunit may act as a negative regulator of thrombin-mediated platelet activation, whereas data from another study suggest that thrombin-gpIb interaction leads to conformational changes in thrombin that reduce its cleavage of fibrinogen.42,43
The phenotype of a gpIbα knockout mouse has been reported and was similar in many ways to human Bernard-Soulier syndrome.44 This mouse was then capable of having the wild-type phenotype restored by a human gpIbα transgene. Future use of this mouse model might be helpful toward further elucidation of the physiologic role of platelet gpIb complex interaction with thrombin.45 In the meantime, the current data extend further support for a role for the gpIb complex as a thrombin receptor on platelets, whereas, recent insights not withstanding, elucidation of the downstream consequences of that interaction with respect to platelet activation and thrombus formation will remain the subject of further investigation.
Studies have focused on interaction of the gpIb complex with activated intact endothelium through ligands other than vWF adherent to subendothelial matrix. These include a study of a reversible association of gpIb with endothelial cell P-selectin, which is examined in more detail in the section, “Platelets and Endothelium.” The interaction of platelet gpIb with the neutrophil adhesion receptor Mac-1 is discussed in the section, “Platelets and White Blood Cells.” These interactions may contribute more to inflammatory responses than platelet thrombus formation. The dependence of fibrin-associated platelet procoagulant activity on both the gpIb complex and vWF has also been documented.46
Glycoprotein Ib Complex Signaling
For some time, controversy surrounded the question of whether the gpIb/V/IX complex signals. This was likely a result of variations in experimental strategies that involve differences with respect to vWF preparations, the use of conformational modulators in experimental work such as botrocetin, cell lines used, and other causes. It is reasonable to assume that the receptor is capable of signaling, although a lot of questions remain to be answered, and the many molecules that are known to participate in the process remain to be assembled into a defined pathway.
When the gpIb complex interacts with its vWF ligand under conditions of elevated shear stress, there is abundant evidence that signaling pathways are activated that lead to (a) elevation of intracellular calcium; (b) activation of a tyrosine kinase signaling
pathway that incorporates nonreceptor tyrosine kinases such as Src, Fyn, Lyn, and Syk, phospholipase C (PlC)γ2, and adaptor proteins such as SHC, LAT, and SLP-76; (c) GTPase activating protein; (d) tyrosine phosphatases (PTP-1B and SHPTP10); (e) inside-out signaling through the αIIbβ3 integrin followed by platelet aggregation32; and (f) activation of protein kinase C (PKC), protein kinase G (PKG), and phosphoinositide 3-kinase (PI3K). vWF binding also up-regulates integrin αIIbβ3 affinity indirectly through stimulation of adenosine diphosphate (ADP) secretion.33 Other downstream players and events that play roles after gpIb receptor occupancy include (a) the homodimeric signaling protein 14-3-3 and calmodulin, (b) receptor cross-linking, and (c) the immunoreceptor tyrosine-based activation motif (ITAM)-containing proteins Fcγ receptor IIA (FcγRIIA) and Fc receptor (FcR) γ chain. In addition to the above-mentioned components, the effect of shear force itself on gpIb signaling, including the affinity and number of bonds between vWF and gpIb, play potentially important roles as well.34,35
pathway that incorporates nonreceptor tyrosine kinases such as Src, Fyn, Lyn, and Syk, phospholipase C (PlC)γ2, and adaptor proteins such as SHC, LAT, and SLP-76; (c) GTPase activating protein; (d) tyrosine phosphatases (PTP-1B and SHPTP10); (e) inside-out signaling through the αIIbβ3 integrin followed by platelet aggregation32; and (f) activation of protein kinase C (PKC), protein kinase G (PKG), and phosphoinositide 3-kinase (PI3K). vWF binding also up-regulates integrin αIIbβ3 affinity indirectly through stimulation of adenosine diphosphate (ADP) secretion.33 Other downstream players and events that play roles after gpIb receptor occupancy include (a) the homodimeric signaling protein 14-3-3 and calmodulin, (b) receptor cross-linking, and (c) the immunoreceptor tyrosine-based activation motif (ITAM)-containing proteins Fcγ receptor IIA (FcγRIIA) and Fc receptor (FcR) γ chain. In addition to the above-mentioned components, the effect of shear force itself on gpIb signaling, including the affinity and number of bonds between vWF and gpIb, play potentially important roles as well.34,35
Evidence has also been presented suggesting that indirect mechanisms may also be involved in gpIb signaling, that are linked to ADP release and/or thromboxane A2 (TxA2) generation.33,34 The increase in intracellular calcium prompted by vWF-gpIb interaction promotes dense granule secretion of ADP, which then activates integrin αIIbβ3 through the P2Y1 and P2Y12 purinergic receptors (see the section on “Platelet P2Y1 and P2Y12 Receptor Roles in ADP-Mediated Activation” later in the chapter).
14-3-3ζ is among the 10 isoforms of the 14-3-3 family of proteins that is named according to its electrophoretic gel migration position. Functionally, this family has a wide range of activities, including participating as a DNA damage, cell-cycle checkpoint protein, regulation of PKC activity, formation of heterotrimers with the signaling kinase Raf and the guanosine triphosphate exchange factor Ras, and so forth.47,48 and 49 Binding sites for 14-3-3 have been identified in the cytoplasmic domains of gpIbα, gpIbβ, gpIX, and gpV. In this respect, peptide fragments corresponding to overlapping cytoplasmic sequences of the four subunits demonstrated binding in vitro, whereas yeast two-hybrid studies documented in vivo interaction between 14-3-3ζ and both gpIbα and –β.50,51 Site-directed mutagenesis and protein-binding experiments confirmed the need for phosphorylation of Ser 166 of the 14-3-3 consensus sequence in the gpIbβ cytoplasmic domain to permit high-affinity 14-3-3 binding.51,52 Findings that have evolved in lieu of the above observations include the binding of fibrinogen to mammalian-cell-transfected αIIbβ3 in response to vWF binding to co-transfected gpIb-IX that was inhibited when the 14-3-3-binding domain was deleted from the gpIb-IX transfectant.53 Also, the lipid kinase PI3K has been found to play a role in this gpIb-IX dependence of αIIbβ3 activation through its participation in complex formation with the gpIb complex and 14-3-3.54 The reason for these associations would be to regulate, in short order, the formation of inositols phosphorylated in the 3-position within the reorganizing cytoskeleton in response to platelet activation (see section “Role of the Cytoskeleton in Platelet Function”).32
Many adhesion receptors initiate signaling through cross-linking, and there is evidence to suggest that the gpIb complex may also use this mechanism. The gpV subunit surface expression on platelets is roughly half that of the other three subunits (12,000 vs. 25,000 copies per cell). It has also been suggested that two or more gpIbα subunits cluster into a complex with the other glycoprotein subunits.55 The fact that there is one gpV subunit available in this complex for every two of the other subunits, and that both actin-binding protein (ABP or filamin; a membrane skeleton protein that associates with gpIbα; see later in the chapter) and 14-3-3 exist as dimers, lends support to the concept of a complex consisting of a pair of gpIbα-gpIbβ-gpIX trimers joined noncovalently by a single gpV monomer (Fig. 17.1).
Several studies document a physical association of the gpIb complex with the 14-kDa FcRγ-chain56 and with the 40-kDa FcγRIIA receptor,57 suggesting that platelet activation may also occur through gpIb-mediated triggering of Src-family-kinase phosphorylation.58,59 Both of these proteins contain nonidentical but similar ITAM domain sequences. The FcRγ-chain forms a complex with the gpVI monomer and plays an essential role in collagen-mediated platelet activation, as reviewed in the next section. The FcγRIIA receptor is a member of the immunoglobulin (Ig) superfamily and is without a clearly defined physiologic role in platelets. Occupancy of this receptor by the C-terminal Fc domains of immunoglobulin leads to platelet activation, and this is blocked by anti-gpIbα antibodies, whereas signaling through vWF is also blocked with anti-FcγRIIA antibodies.57,60 Both ITAM-containing receptors can be coimmunoprecipitated with gpIb,56,60 and both have very similar signaling pathways that involve the tyrosine kinase Syk and PlCγ2.61,62 and 63 Although the physical proximities of these three players have recently become apparent, the nature and significance of any physiologic relationships that exist between the gpIb complex and these two ITAM receptors with respect to vWF-dependent platelet activation remains unclear.
Shear flow studies have demonstrated an important role for calcium signals that in turn promote αIIbβ3 activation necessary for stable platelet adhesion to vWF (34,35). This phenomenon occurs independently of extracellular calcium and is thought to involve αIIbβ3-mediated calcium release from intracellular stores. One proposed mechanism to explain this observation involves signaling pathways such as those used by ITAM-bearing receptors, because the enzyme and adaptor protein components used in both αIIbβ3 and ITAM signaling are very similar. These components are likely to be important in the positive feedback loop linking αIIbβ3 activation and calcium flux under high shear, which in turn promotes the efficient conversion of translocating platelets into firmly adhesive cells.64
Once vWF binds to gpIb-V-IX, signaling complexes form in the vicinity of the gpIbα cytoplasmic tail consisting of cytoskeletal proteins such as 14-3-3ζ as well as signaling proteins such as Src and PI 3-kinase. This process leads to Syk activation, protein tyrosine phosphorylation, and recruitment of other cytoplasmic proteins with Pleckstrin homology domains that can support interactions with 3-phosphorylated phosphoinositides.65,66,67,68 and 69
Platelet-Collagen Interaction and Signaling
Collagens are very important activators of platelets in the vascular subendothelium and vessel wall, and thus are prime targets for therapeutic intervention in patients experiencing a pathologic arterial thrombotic event such as MI or stroke. Platelets have two major surface receptors for collagen, the integrin α2β1 and the immunoglobulin superfamily member gpVI. In addition to binding collagen with high affinity, α2β1 binds laminins, E-cadherins, matrix metalloproteins, C1q, echovirus, and rotavirus. In addition to these two surface receptors, the gpIb complex can also be considered an indirect collagen receptor because its subendothelial vWF ligand essentially acts as a bridging molecule between platelets and collagen by fixing itself to the latter, which, in turn, acts as scaffolding for the multimers.
Collagen supports platelet adhesion through interaction with the integrin surface receptor α2β1, although this interaction alone does not support platelet activation.72 Laboratory evidence suggests that gpVI and the 14-kDa FcRγ-chain signaling subunit (FcRγ chain) with which gpVI forms a complex are both required for collagen-mediated platelet adhesion and activation.61,69,70,71,72,73,74,75,76,77,78,79,80 and 81 Thus platelet adhesion to collagen is a multistep process. Once exposed to an injured vascular wall, vWF adheres to subendothelial collagen and undergoes a conformational change allowing its A1 domain to bind to GPIb/V/IX under high-shear flow conditions. This rapidly formed bond is quickly broken and re-established and this leads to the platelet rolling along the vascular wall. This
in turn slows down the platelet and allows the signaling receptor GPVI to bind collagen, more specifically the repeating sequence Gly-Pro-Hyp (where Hyp is hydroxyproline). Both receptors can signal but the role of GPVI is considered predominant in this respect.82 A series of signaling events then leads to the inside-out signaling activation of α2β1 and interaction of this receptor with collagen is stable and allows the platelet to form a high-affinity strong adhesive bond with the vessel wall.
in turn slows down the platelet and allows the signaling receptor GPVI to bind collagen, more specifically the repeating sequence Gly-Pro-Hyp (where Hyp is hydroxyproline). Both receptors can signal but the role of GPVI is considered predominant in this respect.82 A series of signaling events then leads to the inside-out signaling activation of α2β1 and interaction of this receptor with collagen is stable and allows the platelet to form a high-affinity strong adhesive bond with the vessel wall.
The above notwithstanding, of many receptors studied only α2β1 and GPVI have a defined role in platelet-collagen interactions although, in spite of extensive studies, their relative importance with respect to both adhesion and activation continues to be debatable. Problems associated with the isolation of collagen from extracellular matrices and possible but poorly understood differences between human ex vivo and mouse in vivo experimental systems explain these ongoing unresolved issues.82
α2β1 Receptor
The platelet collagen receptor to be first identified and characterized was the integrin α2β1 receptor, also known as gpIa/IIa, and on lymphocytes as VLA-2.83 Integrins are a family of α–β heterodimers on the surface of cells that carry out diverse interactions between the cell surface and its environment that ultimately lead to changes in cell behavior in response to the ligand-receptor interaction. In all α subunits of integrins, seven tandem repeats are localized to the N-terminal end and folded into a seven-bladed β-propeller structure.84 The α2 subunit also contains an I domain between the second and third repeats that includes a metal coordination site for Mg2+ that is critical for interaction with collagen.85 Similar domains are found on the β subunit, although less is known about these, and it appears that the interaction of α2β1 with collagen involves only the I domain of α2. The β1 subunit exhibits a cysteine-rich domain containing CGXC sequences that is close to the membrane surface. This region has protein disulfide isomerase activity responsible for regulating conformational changes of the β1 subunit (which, in turn, alters α2 conformation, increasing its avidity for collagen) in response to inside-out signaling through the cytoplasmic domain.86,87
Glycoprotein VI Receptor
Although the gpVI receptor was identified on the surface of platelets in 1982, its role in collagen-mediated platelet activation was not appreciated until much later.88 The human and murine genes were cloned and characterized, and gpVI was found to be a member of the immunoglobulin superfamily.81,87,89,90 The expression of gpVI in platelets is very closely associated with that of the 14-kDa FcRγ-chain, which also serves as the signaling subunit for gpVI.91,92 Expression of gpVI on mouse platelets appears to be dependent on FcRγ-chain expression, and the latter has also been found to be critical for collagen-mediated platelet activation.76,92 gpVI has two Ig C2 loops, and the N-terminal loop likely contains the collagen-binding domain.82 It appears that gpVI has a requirement for the quaternary structure of collagen to be in a triple-helical conformation for the two to associate.93
Following the cloning of gpVI, mouse knockout studies have suggested GPVI may be the primary receptor involved in collagenmediated platelet activation.94,95 and 96 Further studies have enabled the collagen-binding site on gpVI to be characterized.97
Studies incorporating collagen-related peptides (arranged in triple helical structures with sequences similar to collagen) and the snake venom convulxin as agonists have been shown to signal by clustering gpVI molecules on the surface.73,78,98 However, the idea of gpVI receptor clustering as a platelet activation mechanism applicable to collagen is tempered by consideration of the theoretically much greater distances that would exist between adjacent gpVI-binding sites along fibrillar collagen compared with the larger, noncovalently linked structures of convulxin, in which gpVI-binding site distribution is much different.99
Platelet-Collagen Signaling
When compared to vWF, collagen is a more efficient substrate when it comes to supporting stable platelet adhesion and thrombus formation. The fact that initial platelet tethering to collagen under high-shear flow first requires interaction between vWF and platelet gpIb serves to underscore the importance of the two major collagen receptors, α2β1 and gpVI, in promoting platelet adhesion and activation under shear conditions.
Many of the early signaling events that follow gpVI stimulation have been characterized, although synergism between these gpVI mediators and those related to other adhesion receptors such as gpIb-V-IX and soluble agonists released by activated platelets further complicates full elucidation of the players and pathways associated with platelet-collagen signaling.
The gpVI signaling pathway has been found to be essential for collagen-mediated platelet activation, and mouse platelet studies have shown a central role for this receptor in promoting platelet-collagen interaction.61,100 Integrin α2β1 also contributes to platelet adhesion through amplification of signals from gpVI.101,102 As noted in the last section, exposure of platelets to collagen surfaces is thought to result in gpVI clustering that in turn triggers the tyrosine phosphorylation of the FcRγ chain.91 gpVI signaling may be influenced by its association with glycolipid-enriched microdomains (membrane rafts) in the plasma membrane, although it is unclear whether gpVI is constitutively associated with the rafts or is recruited.103,104 The gpVI/FcRγ-chain complex leads to platelet activation through a pathway that has many aspects in common with signaling by immune receptors, such as the FcR receptor family (of which FcγRIIA is the lone family member found in platelets) and the B- and T-cell antigen receptors (Fig. 17.2). Much of what we know about gpVI signaling has been based on earlier work related to immune-receptor signaling.105,106
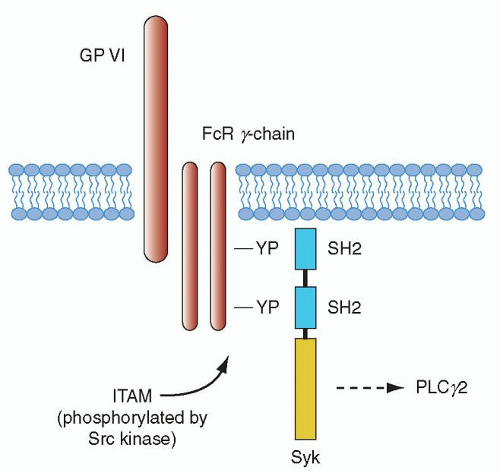 FIGURE 17.2. Collagen activates platelets through the same pathway as an immune receptor. Cross-linking of the glycoprotein (gp) VI/Fc receptor (R) γ chain leads to tyrosine phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM) sequence, enabling recruitment of the tyrosine kinase (Syk) through its tandem Src-homology 2 (SH2) domains. This leads to autophosphorylation and subsequent activation of Syk, which in turn leads to tyrosine phosphorylation of phospholipase Cγ2 (PLCγ2). A large number of proteins are implicated in the regulation of PLCγ2, as discussed in the text. YP, phosphotyrosine. From Watson SP. Collagen receptor signalling in platelets and megakaryocytes. Thromb Haemost 1999;82:367, with permission. |
Immunoreceptors such as the FcRs and the FcRγ chain all have the ITAM in common. Tyrosine phosphorylation of the ITAM by Lyn and Fyn of the Src family of tyrosine kinases takes place after activation of gpVI. The phosphorylated Src kinase, in turn, leads to activation of the tyrosine kinase Syk after its autophosphorylation.107 Syk then initiates a downstream signaling cascade involving the LAT and SLP-76 adapter proteins, which leads to formation of a signaling complex by virtue of its multiple phosphorylation sites that also act as docking sites, leading to recruitment of additional proteins to the plasma membrane.108 Transport to the signaling complex and activation of the cytosolic second messenger-producing enzymes PI3K and PlCγ2 is also facilitated by tyrosine phosphorylated LAT.109 PI3K leads to the generation of PI3,4P3 and PI3,4,5P3 (PIP3), and this, in turn, supports recruitment of proteins to the membrane signalosome complex with specific plekstrin homology (PH) domains including a member of the Tec kinase family, Btk, along with PlCγ2.110,111 Syk is critical for collagen-mediated platelet activation through the gpVI/FcRγ-chain complex, and knockout mouse studies have shown that absence of this enzyme leads to loss of phosphorylation of LAT, the adapter SLP-76, and PlCγ2.61,111,112
Adapters such as LAT are modular proteins without enzyme activity that support protein-protein interaction. Many adapter proteins appear to participate in the regulation of PlCγ2. These proteins appear to come together in T lymphocytes along with PlCγ1 to form a LAT/SLP-76 signalosome that is essential for activation of PlCγ2.106 SLP-76 is thought to be especially important among adapters in the regulation of PlCγ2, because its loss results in reduced phosphorylation of PLCγ2.112,113
Along with the adapter proteins noted above, PI3K and its associated second messenger pathway also play a very important role in the regulation of PlCγ2 through activation of other signaling proteins such as protein kinase B (PKB) (also known as Akt).114,115 One of the two PKB isoforms, PKBβ (Akt2), is important for normal platelet function and thrombus formation and leads to impaired alpha and dense granule secretion along with impaired αIIbβ3 activation when absent.115 Integrin-linked kinase (ILK) is another PI3K downstream effector that interacts with β1 and β3 integrin subunit cytoplasmic tails and is considered important in the bidirectional signaling of α2β1 and αIIbβ3.396,397 It may also be playing a role in regulating PKB.394 Using PI3K inhibitors such as wortmannin and LY294002, studies have demonstrated significantly reduced PlCγ2 activation through gpVI.116,117 and 118 These inhibitors have also been shown to block platelet activation through the FcγRIIA immunoreceptor, and this demonstrates an additional similarity between signaling pathways of this receptor and the FcRγ chain.119
PlCγ2 is known to play a critical role in aggregation and secretion responses to collagen, as demonstrated in PlCγ2 knockout mice and in other studies.63,120,121 The major role of PlC isoforms is concerned with the generation of the second messengers inositol 1,4,5-triphosphate and 1,2-diacylglycerol (DAG), which participate in intracellular calcium homeostasis and activation of many isoforms of PKC. The former process culminates in a robust calcium signal that promotes efficient platelet activation. The PKC isoforms make possible the regulatory serine/threonine phosphorylation events needed for activation of α2β1 as well as αIIbβ3, and interaction of these two integrins with their ligands facilitates a second round of signaling that includes some of the same molecules downstream of gpVI, such as Syk, SLP-76, and PlCγ2.122
Recent research has led to increased knowledge about α2β1 receptor signaling. The use of α2β1-selective ligands has demonstrated calcium-dependent spreading and tyrosine phosphorylation of several proteins when interaction with platelets takes place, including Src, Syk, SLP-76, PLCγ2, p38 MAP kinase, ILK, Rac, and PAK.117,123,124 It appears likely that only GPVI and GPIb/V/IX are able to bind collagen and vWF, respectively, without prior platelet activation. Once activation starts, α2β1 and αIIbβ3 are able to bind their respective ligands as well.
Other Platelet Adhesion Receptors
Other adhesive proteins present in the extracellular matrix and involved in the interaction between platelets and the subendothelium include fibronectin, thrombospondin, laminin, and vitronectin. Fibronectin is stored in platelet α granules and secreted upon thrombin-mediated platelet activation. Thrombospondin is also stored in platelet α granules. It interacts with fibrinogen, fibrin, fibronectin, and collagen on the platelet membrane and its release during platelet activation might relate to its capability to overcome the antithrombotic activity of physiologic nitric oxide (NO). Laminin is a large glycoprotein and can amplify platelet activation through GPVI binding. Recent data suggest that laminin may also interact with vWF and the GPIb/V/IX complex thereby supporting platelet adhesion under high-shear flow conditions.125 The extracellular adhesive protein vitronectin can bind to platelet receptor αIIbβ3 or the integrin αvβ3 and appears to be functionally similar to fibronectin.
Physiologic Inhibition of Platelet Adhesion
Negative regulation of platelets is essential to set the stimulus threshold for thrombus formation, determine final clot size and stability, and prevent uncontrolled thrombosis. The mechanisms behind the negative regulation of platelet activation are described later in the chapter, and in this respect, roles of players such as NO and prostacyclin have been well characterized. Platelet activation and aggregation can also be inhibited by signaling through the platelet endothelial cell adhesion molecule (PECAM)-1 (CD31).126,127 This molecule is expressed on a number of blood cells and endothelial cells and has a wide array of regulatory functions in processes such as apoptosis and cell activation. It becomes tyrosine phosphorylated following platelet stimulation by a diverse set of agonists, which suggests that it has a negative feedback role in this setting. Its main ligand is itself, so it has been proposed that interactions between platelet and endothelial PECAM-1 might serve to restrict thrombus growth through the signaling mechanisms below; this is supported by studies involving PECAM-1-knockout mice.128,129
Following homophilic interactions and/or clustering, PECAM-1 is tyrosine phosphorylated in its cytoplasmic-tail ITIM domain (immunoreceptor tyrosine-based inhibition motif) by Src family kinases, and this engenders recruitment of tyrosine, serine/threonine, or lipid phosphatases with resultant kinase-dependent signaling inhibition.127,130 The net result is reduced total platelet tyrosine phosphorylation, calcium mobilization, and signaling through PI3K.127 ITIM-mediated signaling inhibition through the ITAM domain is not the only mechanism by which PECAM-1 dampens platelet adhesion, because low-density lipoprotein and thrombin-dependent signaling pathways also appear to be downregulated by PECAM-1.127,130 Negative effects have also been documented with gpIb signaling and platelet FcγRIIA-mediated responses.131,132
Von Willebrand factor multimer size and thus also vWF activity is mainly regulated by the metalloprotease ADAMTS13 and recently other factors have been found to cleave vWF including plasmin and leukocyte proteases.133,134 ADAMTS13 cleaves released large vWF multimers into smaller fragments. Different circumstances can induce vWF unfolding thereby exposing the ADAMTS13 cleavage site. These include high-shear flow conditions, denaturing agents such as urea, and mutations seen in von Willebrand disease type 2A. In contrast, reduced ADAMTS13 activity may lead to insufficient vWF processing causing a prothrombotic state such as thrombotic thrombocytopenic purpura (see Chapter 48).
Platelet Thrombin (Protease-activated) Receptors and Signaling
PARs are G-protein-coupled receptors that use a unique mechanism to convert an extracellular protein cleavage event into an intracellular activation signal. In this case, the ligand is already part of the receptor per se, by virtue of the fact that it is represented by the amino acid sequence SFLLRN (residues 42 through 47) and is unmasked as a new amino terminus after thrombin cleaves the peptide bond between Arg 41 and Ser 42. This “tethered ligand” then proceeds to dock irreversibly with the body of its own receptor to effect transmembrane signaling, as shown in Figure 17.3.
Thrombin signaling in platelets is mediated, at least in part, by four members of a family of G-protein-coupled PARs (PAR-1, -2, -3, and -4; see previous section for a discussion of the gpIb complex as a thrombin receptor).135 Human platelets express PAR-1 and PAR-4, and activation of either is sufficient to trigger platelet aggregation.136,137 Mouse platelets express PAR-3 and PAR-4.138 PAR-1, -3, and -4 can be activated by thrombin, whereas PAR-2 can be activated by trypsin, tryptase, and coagulation factors VIIa and Xa but not thrombin.139,140 and 141 Presumably, other proteases are capable of recognizing the active sites of these receptors and can thus also trigger PAR signaling.
PAR-1 is the prototype family member and was the first to be cloned and characterized in the human and hamster.136,142 A synthetic peptide that mimics the PAR-1-tethered ligand (SFLLRN) is capable of functioning as an agonist by activating the receptor independent of cleavage of the 41-residue N-terminal exodomain.
The mechanism by which G-protein-coupled receptors, such as PAR-1, signal through the G proteins is shown in Figure 17.4. PAR-1 is capable of coupling to members of the G12/13, Gq, and Gi/z families and thus is connected to a significant number of intracellular signaling pathways. The α subunits of G12 and G13 are believed to be involved in mediating rearrangement of the actin cytoskeleton and platelet shape change,143 and downstream signaling mediators include Rho family members, among others. The α subunit of Gq is needed for platelet secretion and aggregation and participates in activation of PlCβ that leads to calcium mobilization and PKC activation.144,145 The α subunit of Gz is a Gi family member that has been speculated to play an epinephrinelike role in human platelets through inhibition of adenylate cyclase.34,146
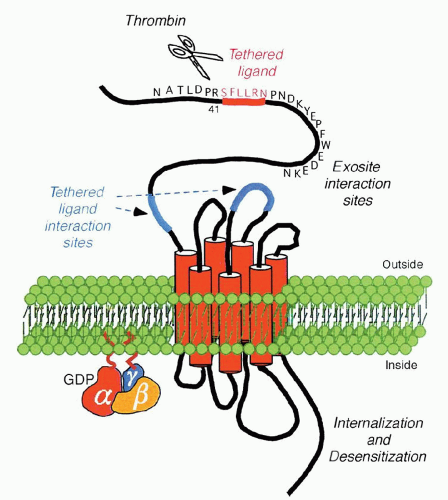 FIGURE 17.3. Structure and features of PAR-1. Cleavage of PAR-1 by thrombin between arginine 41 and serine 42 exposes a new N terminus that serves as a tethered ligand. Activation of PAR-1 is followed by a rapid burst of signaling before the receptor is desensitized and, in some cases, cleared from the cell surface. From Brass LF. Thrombin and platelet activation. Chest 2003;124:18S-25S, with permission. |
The β–γ subunit counterparts of G proteins involved in PAR-1 signaling are involved in a plethora of activities, including activation of protein kinases, channels, and lipid-modifying enzymes, such as PI3K, which provides attachment for multiple signaling protein complexes close to the inner leaflet of the cell membrane.147,148 and 149 Thus, this vast network of signaling pathways mediated through several G-protein families is in keeping with the pleiotropic roles that thrombin has been shown to exhibit in cellular homeostasis, which extends beyond platelet activation to include endothelial cells, leukocytes, smooth muscle cells, and T lymphocytes, along with physiologic processes such as tissue injury, inflammation, angiogenesis, and embryonic development.45
Once activated, PAR-1 is rapidly uncoupled from signaling and internalized into the cell.150,151 and 152 It is then transported to lysosomes and degraded.151,153,154 and 155 Platelets presumably have no need for a thrombin-receptor recycling mechanism, because once activated, they are irreversibly incorporated into blood clots. Conversely, in cell lines with characteristics similar to megakaryocytes (MKs), new protein synthesis is needed for recovery of PAR-1 signaling151,153 and, in endothelial cells, sensitivity to thrombin is maintained by delivery of naïve PAR-1 to the cell surface from a preformed intracellular pool.153
Physiologic differences exist between PAR-1 and PAR-4 on human platelets. When antibodies to the thrombin interaction site of PAR-1 are used, platelet activation is blocked at low, as opposed to high, thrombin concentrations.154,155 Antibodies that blocked PAR-4 alone had no effect on thrombin-mediated platelet activation. If both receptors were blocked, platelet activation was blocked at both low and high thrombin concentrations,138 and so PAR-1 appears to be most efficient at mediating platelet activation at low concentrations of thrombin, whereas PAR-4 functions in the absence of PAR-1 but only at high thrombin concentrations. Because PAR-1 is capable of mediating platelet activation at low thrombin concentrations, the exact role of PAR-4 in human platelet function remains speculative. It has been shown that the rate of platelet activation through PAR-4 is significantly slower and more sustained than that through PAR-1.45,156,157 Given the importance of this system with respect to normal hemostasis, PAR-4 may serve as a redundant backup receptor to PAR-1, or it may serve as an important receptor for one or more proteases other than thrombin.
Initial studies addressing the role of thrombin in promoting platelet thrombus formation employing ex vivo perfusion systems pointed toward its importance at low- to intermediate-shear rates (100 to 650 s-1), but not at higher shear rates (>2,600 s-1).158,159 Subsequent studies employing PAR3 and PAR4 knockout mice have also demonstrated defective thrombus formation under high-shear conditions in vivo, although suggesting the possible existence of shear-based functional differences between PARs.160,161
Platelet ADP (Purinergic) Receptors and Signaling
Evidence that ADP plays an important role in both formation of the platelet plug and the pathogenesis of arterial thrombosis has been accumulating since its initial characterization in 1960 as a factor derived from red blood cells that influences platelet
adhesion.161,162 ADP is present in high (molar) concentrations in platelet-dense granules and is released when platelet stimulation takes place with other agonists, such as collagen; thus, ADP serves to amplify further the biochemical and physiologic changes associated with platelet activation and aggregation. Inhibitors of this ADP-associated aggregation include commonly used clinical agents such as clopidogrel that have proven to be very effective antithrombotic drugs.163,164
adhesion.161,162 ADP is present in high (molar) concentrations in platelet-dense granules and is released when platelet stimulation takes place with other agonists, such as collagen; thus, ADP serves to amplify further the biochemical and physiologic changes associated with platelet activation and aggregation. Inhibitors of this ADP-associated aggregation include commonly used clinical agents such as clopidogrel that have proven to be very effective antithrombotic drugs.163,164
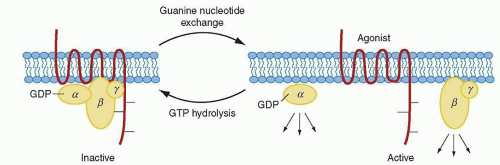 FIGURE 17.4. Signaling through G proteins and G-protein-coupled receptors. Agonist binding to the receptor causes the exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on the guanine nucleotide-binding site of the G-protein α subunit. This causes the dissociation of Gα from Gβγ, both of which can activate intracellular effectors and ion channels. From Brass LF. Molecular basis of platelet activation. In: Hoffman R, Benz EJ, Shattil SJ, et al., eds. Hematology: basic principles and practice. New York: Churchill-Livingstone, 2000:1754, with permission. |
Adenine nucleotides interact with P2 receptors that are ubiquitous among different cell types and have been found to regulate a wide range of physiologic processes. They are divided into two groups, the G-protein-coupled or “metabotropic” superfamily named P2Y and the ligand-gated ion channel or “ionotropic” superfamily termed P2X.165 Two G-protein-coupled (P2Y) receptors contribute to platelet aggregation. The P2Y1 receptor initiates aggregation through mobilization of calcium stores, and the P2Y12 receptor is coupled to inhibition of adenylate cyclase and is essential for a full aggregation response to ADP with stabilization of the platelet plug. P2X1 is a third ADP receptor present in platelets and has been shown to contribute to aggregation in response to collagen. Both the P2Y12 and P2X1 receptors have been shown to play key roles in platelet activation and aggregation under flow conditions characterized by high-shear stress.166,167
The P2Y1 receptor was first cloned in 1993 from a chick brain complementary DNA library.168,169 and 170 Messenger RNA was later found in MK-like cell lines, such as HEL and K562, along with human platelets.171 It was also established that the purported agonist effects of purified triphosphate nucleotides were, in fact, due to their transformation into diphosphate analogs by the ectonucleotidases present at the cell surface of the platelets and brain capillary endothelial cells being studied.171,172 The P2Y1 receptor has 373 amino acid residues and the prototype structure of a G-protein-coupled receptor. It is distributed in various tissues such as heart, blood vessels, testis, and ovary.165
After the characterization of P2Y1, it became clear that a second platelet ADP receptor had to exist which was responsible for the inhibition of cAMP production by ADP that, in turn, was unaffected by blocking P2Y1.173,174,175,176,177 and 178 The P2Y12 receptor was cloned in 2001 from human and rat platelet complementary DNA libraries using Xenopus oocytes.179 The receptor indeed showed the ability to display ADP-mediated inhibition of platelet cyclic AMP (cAMP) formation that was not blocked by P2Y1 antagonists. The receptor has been localized to certain regions of the brain, such as the substantia nigra and thalamus, in addition to platelets.179
Platelet P2Y1 and P2Y12 Receptor Roles in ADP-Mediated Activation
Even at high concentrations ADP is a weak activator of PlC. Thus its role in platelet activation is based more on its ability to activate other pathways. Inhibition of either of the P2Y1 or P2Y12 receptors is sufficient to block ADP-mediated platelet aggregation, and co-activation of both receptors is therefore necessary, through the G proteins Gq and Gi, respectively, for ADP to activate and aggregate the platelet (Fig. 17.5).180 A series of studies involving the use of selective P2Y1 and P2Y12 receptor antagonists, a cAMP inhibitor, gene targeting, and Gq and Gi protein agonists that would theoretically activate the two main G-protein pathways associated with ADP stimulation (see below) has led to the conclusion that a signaling event downstream from Gi is required for the conformational change and subsequent aggregation associated with the αIIbβ3 receptor.176,178,180,181,182,183,184,185 and 186
Studies done with platelets from patients who manifest defective P2Y12, along with experiments involving the study of P2Y1 receptor function in platelet-rich plasma that has high fibrinogen concentrations, have demonstrated that the P2Y1 receptor has roles in activation and aggregation in addition to shape change, and that it is fully capable of mounting an aggregation response which is nonetheless transient in nature.176,187,188 The P2Y1 receptor is an absolute requirement for ADP-mediated aggregation based on knockout mouse studies, the demonstration that platelets can become refractory to ADP due to desensitization of the P2Y1 receptor, and the observation that adrenaline (which activates the G protein Gi that mediates inhibition of adenylate cyclase) does not restore aggregation in the presence of P2Y1 selective antagonists.183,184,185,186,187 and 188 Platelet shape change is dependent on two separate G signaling pathways, a Gq-linked release of calcium from internal stores, and a G12/G13 link to activation of Rho kinases and Rho guanine nucleotide exchange factors that activate small G proteins.143,189,190 The primary role of the P2Y12 receptor in platelet activation and aggregation is to amplify and complete the aggregation response to ADP as well as to other agonists.173,174,175,176 and 177 In the presence of a high ADP concentration, the receptor is also capable of mediating partial platelet aggregation on its own in P2Y1 and Gq knockout mice, thus proving that αIIbβ3 conformational change followed by aggregation can actually take place in the absence of calcium mobilization and PKC activation.184,190 In general terms, it appears that P2Y12 is responsible for acting as an ADP co-stimulus receptor in the presence of low concentrations of other agonists, such as collagen, thrombin, chemokines, or IgG, whereas the P2Y1 receptor has a specific role early in platelet activation.191,192 and 193 Another role for the P2Y12 receptor is the potentiation of platelet secretion.194,195 Because of its central role in the formation and stabilization of thrombi, the P2Y12 receptor is a well-established target of antithrombotic drugs such as clopidogrel.194,196
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


