FIGURE 6-1. Modular structure of nuclear hormone receptors. The most highly conserved domain is the DNA-binding domain, DBD, followed by the ligand-binding domain, LBD. The domains are sometimes also referred to by their alphabetic designations. Functions of the domains are indicated. The N-terminal or A/B domain is highly variable in length and sequence and is not present in some receptors. The F domain is present in only a limited number of receptors and is also not conserved in sequence. Its functions have generally not been well characterized, but it is thought to modulate transactivation in some cases.
A more limited number of receptors have short C-terminal extensions (F domain) after the LBD. These are often dispensable for basic transcriptional regulation but may have modulatory functions.
DNA-BINDING DOMAINS
The DNA-binding sites recognized by the receptors are called hormone response elements (HREs), and are present in promoters and regulatory regions of receptor target genes. Nuclear hormone receptors bind three different types of response elements. Except for the estrogen receptors (ERs), which can heterodimerize with each other, the steroid receptors function as homodimers and recognize two copies of a hexameric sequence related to the consensus 5′ AGAACA 3′, which are separated by 3 base pairs and arranged as a head-to-head inverted repeat. More than a dozen of the other nuclear receptor family members bind DNA as heterodimers with the RXRs; nearly all are either classic receptors (TRs, RARs, VDR) or new receptors (PPARs, LXRs, FXR, CAR, PXR). Surprisingly, the RXRs, their heterodimer partners, the ERs, and nearly all of the orphan receptors recognize hexameric motifs related to a consensus, 5′ AGGTCA 3′, which is similar to that bound by the other steroid receptors. Most of these complexes bind as dimers, but several orphan receptors can bind the same hexameric consensus element as monomers. In this mode, a C-terminal extension of the DBD makes additional base-specific contacts upstream of the hexamer, with different receptors recognizing different sequences. The structures of homodimeric, heterodimeric, and monomeric receptor–DNA complexes are shown in Fig. 6-2A.
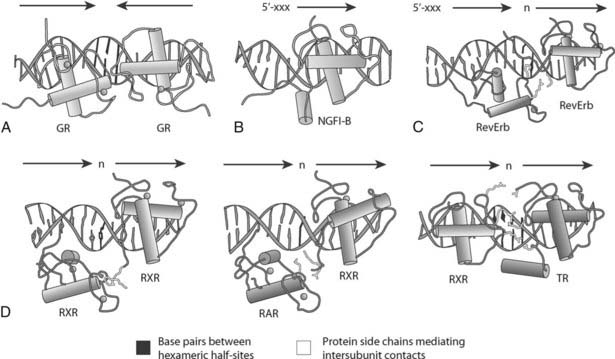
FIGURE 6-2. Structures of complexes between receptor DNA-binding domains and their cognate DNA-response elements. A, GR homodimer bound to an inverted element with a 3-base-pair spacer (IR-3). B, NGFI-B bound to its extended monomeric site. C, RevErb homodimer bound to an extended direct repeat element with a 2-base-pair spacer (DR-2). D, RXR as a homodimer bound to a DR-1 element, RAR/RXR on a DR-1 element, and TR/RXR heterodimer bound to a DR-4 site. Note that RXR binds only at the upstream half-site on the DR-4 HRE with TR, and only at the downstream half-site on the DR-1 with RAR. Cylinders indicate helices, base pairs between the hexameric half-sites are shown in red, and protein side chains mediating intersubunit contacts are shown in yellow.
(Reproduced with permission from Khorasanizadeh S, Rastinejad F: Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci 26:384–390, 2001.)
The ability of the vast majority of the receptors to recognize the AGGTCA consensus creates an obvious specificity problem that is addressed in part by variations in the spacing and arrangement of the two hexameric binding sites. For example, TR/RXR heterodimers recognize direct repeats of this hexamer separated by 4 base pairs, while RAR/RXR and VDR/RXR heterodimers prefer 5-base-pair and 3-base-pair spacers, respectively.50 These rules are not absolute, since the receptor complexes are remarkably flexible and can often bind multiple types of elements. TR/RXR complexes, for example, can also bind head-to-head inverted repeats of the hexamer with no spacer, as well as tail-to-tail or everted repeats separated by 6 base pairs. Based on the diversity of sites for a single receptor complex and the large number of receptors, it is not surprising that a particular element can often be recognized by multiple receptor complexes. Although they are not well defined, additional mechanisms such as cell- and tissue-specific expression of receptors and coregulators, as well as differential interactions with other transcription factors, must allow specific receptors to appropriately regulate their target genes.
High-resolution x-ray crystal structures have been solved for complexes of DBDs with their response elements. The DBDs are primarily α-helical, compact units of 66 to 68 amino acids that fold around two Zn++ ions, each of which is coordinated by four invariant cysteine residues. The receptor DBDs are frequently described as “zinc fingers,” but this is not strictly accurate, since they are folded together in a single unit and are not functionally independent.
As shown in Fig. 6-2A and recently reviewed,51 the structures of appropriate DBDs and response elements provide detailed information on the distinct modes of binding. For the homodimeric steroid receptors, the structures reveal specific head-to-head protein-protein contacts that lock the two DBDs in position to bind the two inverted hexamers.52 Since the hexamers are separated by approximately one turn of the double helix, the two monomers bind the same face of the helix. A similar mode of binding is evident from the structures of some of the RXR complexes, but different head-to-tail contacts with RXR position the various partners appropriately and allow the complexes to recognize different direct repeat response elements.53
Like many other transcription factors, specific DNA contacts are made by residues present in short α helices. The highly conserved but distinct sequences of the primary recognition helix, termed the P-box,54 account for the ability of the steroid receptors and the other members of the superfamily to bind the two distinct hexameric consensus sites. The receptors that bind as monomers also use two helices to make specific DNA contacts, with the additional helix coming from the C-terminal extension of the conserved DBD.55
LIGAND-BINDING DOMAINS
In addition to binding ligand, LBDs function in receptor dimerization and transcriptional activation. The molecular mechanisms for all of these functions have been revealed by numerous x-ray crystal structures of LBDs, with or without their cognate ligands or coregulator peptides. Despite a low degree of primary sequence conservation across the superfamily, the overall LBD structure is highly conserved and is typically described as an antiparallel three-layered sandwich of 12 α helices. In the conventional receptors, a portion of the middle layer is missing, creating a pocket for the ligand. By convention, this is considered the lower portion of the structure.
The structures of the ligand-occupied steroid, thyroid hormone, and retinoic acid receptors are quite consistent with the high affinity and specificity of their ligands. The hormone fits very tightly into the pocket, making multiple favorable contacts with the residues that line it. Importantly, ligand binding results in appropriate positioning of the C-terminal helix 12, which forms a hydrophobic cleft with portions of helices 3, 4, and 5. This surface is the binding site for the large number of transcriptional coactivators that mediate nuclear receptor transactivation of gene expression, and allosteric modulation of the structure of helix 12 is the molecular mechanism for the modulation of the AF-2 transcriptional activation function of the LBD by ligand (Fig. 6-3).
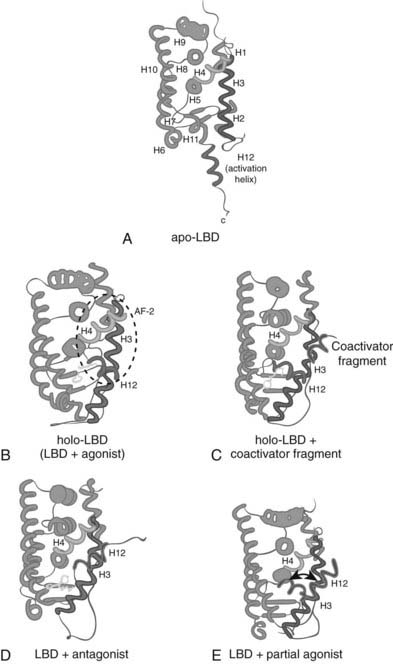
FIGURE 6-3. Diagrams of LBDs of representative NRs. Protein, green; helix 3, blue; helix 4, pink; ligands, yellow; helix 12, red; LXXLL motif of coactivators, violet. Helices are numbered 1 to 12 as reported for the first NR structure, RXR. A, The apo-form of RXR, the binding site of which is not accessible to ligand (PDB entry code 1lbd). B, The binary complex of RAR and all-trans retinoic acid in the transcriptionally active form (PDB entry code 2lbd). C, The ternary complex of ER, distilbestrol, and a fragment of the coactivator GRIP, which contains the LXXLL motif (PDB entry code 3erd). This structure represents the transcriptionally active form of NRs and indicates the binding site of coactivators. D, The binary complex of ER and the selective ER modulator tamoxifen (PDB entry code 3ert). This structure represents a transcriptionally inactive form of NRs where helix 12 is located in the binding site of coactivators. E, The binary complex of ER and the partial agonist genistein (PDB entry code 1qkm). In the crystal structure, helix 12 is located in the coactivator-binding groove. Helix 12 as observed in the transcriptionally active form of NRs is superimposed to illustrate the alternative positioning of helix 12 in complexes of NRs with partial agonists as underlined by the double pointed arrow.
(Reproduced with permission from Steinmetz AC, Renaud JP, Moras D: Binding of ligands and activation of transcription by nuclear receptors. Annu Rev Biophys Biomol Struct 30:329–359, 2001.)
The contact between activated nuclear receptors (NRs) and coactivators is mediated primarily by a remarkably short conserved element found in most coactivators, the LXXLL motif or NRbox.71 The conserved leucine residues of the coactivator NRbox are found on the same face of an amphipathic helix and fit into the hydrophobic cleft. Coactivator binding is also supported by charge-based interactions between conserved receptor residues and the helical backbone of the coactivator motif that are referred to as the charge clamp.71
Agonist ligands stabilize the appropriate position of helix 12 by hydrogen bonding or other direct interactions. In contrast, antagonist ligands force it to adopt alternate conformations that do not allow coactivator binding. In examples such as 4-hydroxytamoxifen, a portion of the antagonist extends into the space where helix 12 would be found in an activated receptor and displaces it.72 This helix is also amphipathic, and the hydrophobic face can fold back onto the receptor surface to occupy the remainder of the coactivator cleft. As with the agonists, antagonists can also disrupt the AF-2 structure by less direct means.
In contrast to this hand-in-glove mode of binding for the classic receptors, the ligand-binding pockets of the new metabolic receptors can be much bigger than the ligands that bind them. For example, eicosapentaenoic acid occupies only a fraction of the ligand pocket of PPARδ, with the acyl side chain adopting two quite different structures that are each supported by weak hydrophobic interactions with distinct residues that line the pocket.14 Based on this, it is not surprising that many different fatty acids can bind PPARδ and the other isoforms with similar affinities, or that ligands such as the synthetic thiazolidinediones that fill the PPARγ pocket more completely bind with much higher affinity and specificity. As with the conventional hormone receptors, both low-affinity and high-affinity agonists for the more promiscuous receptors function to stabilize the active conformation of helix 12.
Crystal structures have revealed unexpected features of the LBDs of orphan receptors. In some cases, unexpected constituents have been observed in the ligand-binding pocket. The fatty acids in the HNF-4α pocket56,73 and the cholesterol in the retinoid-related orphan receptor α (RORα) pocket57 presumably bound to and stabilized them in the Escherichia coli host used to express the crystallized LBDs. Particularly for HNF-4α, studies indicate that the fatty acids are essentially permanent occupants of the cavity that do not modulate receptor function and may be more analogous to the Zn++ atoms in the DBD than to conventional ligands.
In contrast, the potential pocket of Nurr1 is fully occupied by bulky amino acid side chains.76 It has been speculated that this may actually be the primordial LBD structure, particularly since apparently ligand-independent orphan receptors are the most highly conserved superfamily members in distantly related species. The HNF-4α structure suggests that the transition to hormone responsiveness may have begun with the mutation of a bulky side chain in the hydrophobic core to a smaller residue, resulting in a pocket that could have been occupied initially by structural ligands.
Crystal structures also reveal the basis for the dimerization function of the LBDs. Parallel contacts between helix 10 in each monomer form the primary interface, with additional contacts made between helix 7, the loop between helices 8 and 9 on one side, and between helix 9 and the N-terminus of helix 10 on the other. Steroid receptors and RXR homodimers are symmetrical, but this symmetry is slightly disrupted in the RXR heterodimers. A recent analysis suggests that all receptors can be identified as homodimerizing or heterodimerizing based on only a few differentially conserved LBD residues that affect this interface.58 However, the structure of the GR LBD indicates that it uses a quite different dimerization strategy based on interactions between beta sheets.78
New insights into additional higher-order structural features of nuclear receptors have been provided by the recently reported x-ray crystal structure of the intact PPARγ-RXRα heterodimer bound to its hormone response element as well as to ligands for both receptors and also coregulator peptides.59 All of the domains adopt the expected conformations and interactions as outlined above. However, the complex is asymmetrical, and the two LBDs adopt quite different positions (Fig. 6-4). Thus the RXRα LBD is farther from the DNA, while the PPARγ LBD packs between it and the two DBDs. As expected, the N-terminal A/B segments are too flexible to be resolved. It seems that the PPARγ LBD provides the core, interacting with both DBDs to promote correct hormone response–element binding, and also coordinating the overall structure of the complex. It remains to be seen whether the general features of this structure will be broadly applicable to other complexes or if distinct receptors adopt quite different higher-order confirmations.
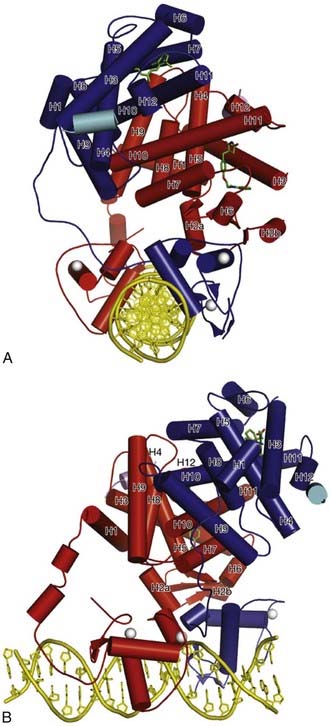
FIGURE 6-4. Crystal structure of PPAR-RXR heterodimer bound to DNA response element. RXRα is shaded dark and PPARγ is shaded lighter. The ligands rosiglitazone and 9-cis-retinoic acid are shown chemical structures, Zn(II) ions are solid balls, and the DNA double helix is shown at the bottom of the figure.
(Reproduced with permission from Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F: Structure of the intact PPAR-gamma-RXR nuclear receptor complex on DNA. Nature 456:350–356, 2008.)
Regulation of Gene Expression by Nuclear Receptors and Coregulators
Nuclear receptor coregulators have been historically divided into coactivators, which exert a positive influence on nuclear receptor–mediated transcription, and corepressors, acting in the opposite direction. These terms arose initially based upon empirical descriptions of the effect of these molecules on what are by today’s standards relatively unsophisticated assay endpoints—the transcriptional output of a minimal reporter, for example. As the characterization of coregulators has progressed to assays more reflective of the complexity of promoters in their native biologic contexts, the functional polarity that these terms imply is coming under increasing scrutiny.60 We will apply this distinction in this section solely for the sake of clarity. The reader should be careful to bear in mind that these are general terms, and increasing evidence indicates that ligand and nuclear receptor identity, in addition to cell and promoter context, are all powerful influences on the biology of a given coregulator at any given point in time.
COACTIVATORS
Soon after the existence of limiting ancillary factors in transcription was first suggested by studies in yeast, evidence for their function in nuclear receptor transactivation was provided by competitive effects between receptors or with receptors and other transcription factors. The demonstration of hormone-dependent recruitment of specific proteins to the activated estrogen receptor60–63 was followed by the isolation of cDNAs encoding ligand receptor–interacting proteins in many laboratories. Functional studies confirmed that such ligand-dependent receptor interactors can act as coactivators to support hormone-dependent transcriptional activation.
Two primary aspects of the multi-step process of transcriptional activation are: (1) counteracting the inherent repressive effects of the packaging of genes into chromatin, and (2) recruiting RNA polymerase and the basal transcriptional apparatus to the promoter. The theme that has emerged is that binding of a ligand-activated receptor to an appropriate target gene results in the recruitment of a surprisingly large number of multi-protein complexes that mediate these effects via a variety of enzymatic activities.
DNA packed into chromatin is obviously less accessible than free DNA, and chromatin plays a dominant repressive role in the basal activity of genes in eukaryotic cells. As shown in Fig. 6-5, recruitment of complexes designed to overcome this constraint is thought to be an early step in receptor-dependent transcriptional activation. Among the best characterized of these are a series of multi-protein complexes that contain one of two ATPase subunits, called BRG1 and brahma, which are related to the yeast protein SNF2. They are referred to as SWI/SNF complexes based on similarities to the complex originally described in yeast. Both the yeast and mammalian complexes use the energy from adenosine triphosphate (ATP) hydrolysis to remodel nucleosomes, rendering them more accessible to transcription factors.63 In at least some cases, their function is essential for nuclear receptor transactivation.64
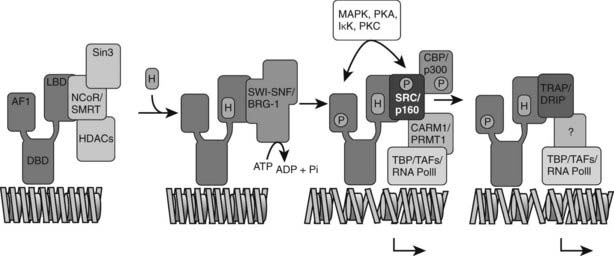
FIGURE 6-5. Model of combinatorial NR-mediated transcriptional initiation. Initial binding of ligand results in dissociation of corepressors and recruitment of SWI/SNF chromatin remodeling machines to modify chromatin domains. Binding of SRCs and CBP results in local acetyltransferase activity and disruption of local nucleosomal structure. Kinase-mediated signaling pathways may communicate directly with NR-regulated promoters. AF-1 phosphorylation might serve to further consolidate ligand-dependent NR-SRC interactions or to recruit SRCs directly to the promoter in the absence of ligand. TRAP/DRIP directly contacts components of the basal transcription machinery to effect transcriptional initiation, and certain TAFs may afford some additional input into promoter-specific NR transcription. The extent of overlap in binding of complexes to the promoter is currently unclear. Local coactivator requirements may vary. For example, a promoter in a readily accessible chromatin context may not require significant chromatin remodeling or histone acetyltransferase activity for assembly of a preinitiation complex.
(Reproduced with permission from McKenna NJ, O’Malley BW: Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474, 2002.)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


