The spectrum of bone marrow failure disorders and other myeloid neoplasms. AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MPN, myeloproliferative neoplasm; CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndromes; PNH, paroxysmal nocturnal hemoglobinuria; LGL, large granular lymphocyte leukemia; ITP, immune thrombocytopenia; PRCA, pure red cell aplasia.
| Name | Peripheral blood findings | Key bone marrow findings |
|---|---|---|
| Chronic myelomonocytic leukemia type 1 (CMML-1) | > 1 × 109/L monocytes for ≥ 3 months <5% blasts | Uni- or multilineage dysplasia, <10% blasts (not including abnormal monocytes) |
| Chronic myelomonocytic leukemia type 2 (CMML-2) | >1 × 109/L monocytes for ≥3 months 5–19% blasts or Auer rods | Uni- or multilineage dysplasia, 10–19% blasts or the presence of Auer rods |
| Myelodysplastic syndrome (MDS)/myeloproliferative neoplasm, unclassifiable (MPN-U) | Dysplasia with myeloproliferative features, no prior diagnosis of MDS or MPN | Dysplasia with myeloproliferative features, but not meeting criteria for MDS, MPN, or other overlap syndrome |
| Refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) (provisional) | Anemia, no blasts, platelets ≥450 × 109/L | ≥15% of erythroid precursors with ring sideroblasts, erythroid dysplasia only, <5% blasts on morph, proliferation of large megakaryocytes, presence of SF3B1 mutation |
Chronic myelomonocytic leukemia
Case 1: part 1
A 72-year-old woman presents to her primary care physician with a 4-month history of fatigue. On review of systems she adds that she has had intermittent left-sided abdominal pain and fullness. Physical examination reveals mild splenomegaly (spleen tip 3 cm below the left costal margin at the mid clavicular line), but is otherwise unremarkable. A review of her blood counts is notable for normocytic anemia (hemoglobin 9.2 g/dL) and a white count of 10.3 × 109/L with monocytosis (1.8 × 109/L). Laboratory work from her annual visit 6 months ago also demonstrated mild anemia (10.3 g/dL) and monocytosis (1.2 × 109/L). She has not required any transfusions.
Clinical presentation
Given the context of this chapter, it would not be surprising to conclude that the woman in this case may have an overlap syndrome. However, it is important to consider alternative diagnoses with the initial presentation, in particular, chronic myeloid leukemia (CML). By definition, CMML is negative for BCR-ABL1 translocations (Figure 14.2), but CML can present with the same constellation of symptoms and lab findings and in the absence of genetic investigation be misdiagnosed. With the discovery of the Philadelphia chromosome and development of imatinib and other tyrosine kinase inhibitors, it is critical that CML is excluded from the differential diagnosis. Entities defined by rearrangements in platelet-derived growth factor receptor (PDGFRα or PDGFRβ) genes also have exquisite imatinib sensitivity and should be ruled out as well. This is emphasized by the fact that CMML and other myeloid neoplasms with PDGFRα/β rearrangements have their own diagnostic category as defined by the WHO: myeloid/lymphoid neoplasia with eosinophilia and abnormalities in PDGFRβ, PDGFRα, or FGFR1. Although much less common, aCML can also present with monocytosis, but by definition monocytes are less than 10% of the peripheral blood differential (see Chapter 15).
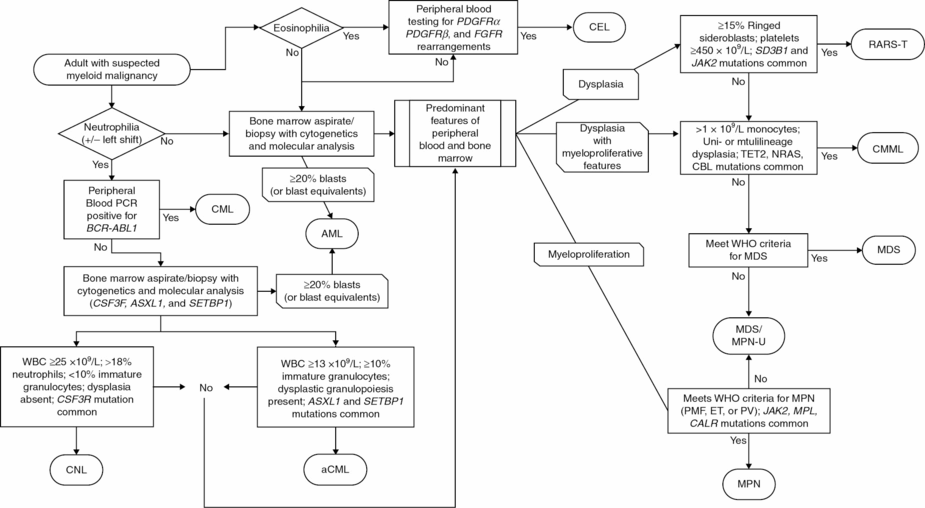
Diagnostic workflow for myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) overlap syndromes.
PMF, primary myelofibrosis; ET, essential thrombocythemia; PV, polycythemia vera; PCR, polymerase chain reaction; WBC, white blood cell; WHO, World Health Organization; blasts, myeloblasts; CEL, chronic eosiniphilic leukemia; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MPN, myeloproliferative neoplasm; CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndromes; CNL, chronic neutrophilic leukemia; aCML, atypical chronic myeloid leukemia; RARS-T, refractory anemia with ringed sideroblasts and thrombocythemia; MDS/MPN-U, MDS/MPN overlap unclassified.
Patients with symptoms suggesting the release of mast cell mediators (i.e., diarrhea, pruritus, intermittent hypotension, nausea, anaphylaxis), or who have lesions suggestive of mast cell infiltration such as urticaria pigmentosa, should be considered for systemic mastocytosis with associated clonal hematological non-mast cell lineage disease (SM-AHNMD). The workup is similar to that of other forms of mastocytosis, including serum tryptase levels, bone marrow biopsy, and molecular testing (see Chapter 12).
In the absence of a distinct genetic marker akin to CML, it is more difficult to draw the line between CMML and MDS. From a practical standpoint this is of less importance, as again the current care of CMML patients is heavily extrapolated from MDS. Nonetheless, precise diagnosis is important as it holds prognostic relevance and allows for the identification of patients for clinical trials and potentially targets in this frequently overlooked disease category.
This case demonstrates the hallmarks of CMML at presentation. The namesake of the disorder is persistent monocytosis, and workups are often triggered by abnormal lab values in the setting of mild constitutional symptoms such as fatigue. It is also important to note that, as in our case, between a quarter and half of patients will present with splenomegaly, a nod to the proliferative half of the disorder.2,3 Other non-medullary symptoms include hepatomegaly, lymphadenopathy, or nodular cutaneous myeloid or leukemic infiltrates (i.e., Sweet’s syndrome).
Case 1: part 2
Given the suspicion of a myeloid neoplasm the patient underwent a bone marrow aspirate and biopsy. The biopsy was hypercellular (70%) and the aspirate revealed 4% blast equivalents (myeloblasts plus monoblasts) and megakaryocyte dysplasia. Metaphase cytogenetics revealed a normal female karyotype. Molecular diagnostics on the marrow sample revealed the absence of a BCR-ABL1 fusion protein, and there were no rearrangements of PDGFRα and PDGFRβ. However, a mutation was found in TET2.
Epidemiology
With the results of this bone marrow biopsy, the patient in the case meets the diagnostic threshold for CMML. The majority of patients are diagnosed in their seventh and eighth decades of life and there is a slight male predominance.4 Although challenges in distinguishing the diagnosis from related disorders and coding lead to underestimation, in 2011 the Surveillance, Epidemiology, and End Results registry reported an age-adjusted incidence of 0.42 per 100,000 per year.5 A population-based study conducted in Spain gauged the incidence to be 0.41 per 100,000 per year, accounting for 90% of all MDS/MPN overlap diagnoses.6
Prognosis
Similar to other malignancies, the treatment of CMML has been traditionally risk-adapted, where the aggressiveness of the treatment is matched to the aggressiveness of the disease. However, there are few data to support this approach. Nonetheless, in a group with heterogeneity of outcome, accurate prognosis is key in making treatment decisions. The original and revised International Prognostic Scoring System (IPSS and IPSS-R) are often used, despite only 15% and 9% of the derivation cohort being comprised of CMML for the IPSS and IPSS-R, respectively.7,8
Addressing this issue, CMML-specific models have been developed using clinical factors common in CMML, and, to no big surprise, are recurrent in prognostic nomograms for MDS and MPN. Originally reported in 2002, the MD Anderson Prognostic Score (MDAPS) has been a mainstay prognostic tool.9 More recently, the CMML-specific prognostic scoring system (CPSS)10 groups patients into risk categories by accounting for the presence of chromosomal abnormalities, red cell transfusion dependence, bone marrow blast count, and leukocytosis (Table 14.2). In validation studies, the CPSS may outperform the MDAPS.11 By the CPSS, the patient in the case would have low-risk disease with a median overall survival of 72 months.
CPSS cytogenetic risk groups
| Cytogenetic prognostic subgroups | Cytogenetic abnormalities |
|---|---|
| Low | Normal, isolated –Y |
| Intermediate | Any other single or double independent clones |
| Poor | +8, chromosome 7 abnormalities, and complex karyotype (≥3 abnormalities) |
CPSS prognostic score values
| Prognostic variable | 0 | 1 | 2 |
| WHO subtype | CMML-1 | CMML-2 | – |
| FAB subtype | CMML-MD (WBC count < 13 × 109/L) | CMML-MP (WBC count ≥ 13 × 109/L) | – |
| Cytogenetic risk | Low | Intermediate | High |
| RBC transfusion dependencya | No | Yes | – |
WHO, World Health Organization; FAB, French, American, and British; CMML-MD, myelodysplastic syndrome-variant CMML; WBC, white blood cell; CMML-MP, myeloproliferative-variant CMML; RBC, red blood cell.
a RBC transfusion dependency was defined as having at least one RBC transfusion every 8 weeks over a period of 4 months.
CPSS prognostic risk categories/scores
| Risk category | Risk score |
|---|---|
| Low | 0 |
| Intermediate-1 | 1 |
| Intermediate-2 | 2–3 |
| Very high | 4–5 |
CPSS prognostic risk category clinical outcomes
| Total | Low | Intermediate-1 | Intermediate-2 | High | |
|---|---|---|---|---|---|
| Patients (%) | 558 | 217 (41) | 155 (29) | 141 (26) | 19 (4) |
| Median survival | 5.1 | 2.6 | 1.1 | 0.4 | |
| Risk of acute myeloid leukemia at 2 years | 8% | 25% | 49% | 100% |
Even with CMML-specific prognostic systems, the prognostic value of existing clinical variables has been maximized, and new methods to describe an individual’s disease risk are needed. The natural next step is to incorporate the ever-growing library of molecularly defined lesions (Figure 14.3). As discussed in Chapter 2, molecular exploration of these diseases has the potential to lift the lid on the apparatuses of malignancy and allow for precise targeting of therapeutics. Currently these mutations that can independently predict survival and disease progression are being incorporated into prognostic models. An analysis of 226 patients conducted by the group at the Mayo Clinic found that that neither ASXL1 nor spliceosome mutations were associated with survival when incorporated with traditional clinical features.12 Conversely, the Groupe Francophone des Myelodysplasies (GFM) genotyped ASXL1 and up to 18 other genes in 312 patients with CMML to derive a prognostic score integrating both clinical and molecular predictors and found that mutations in ASXL1 retained its association with an inferior overall survival (hazard ratio 1.76; 95% confidence interval 1.22–2.53; p < 0.002).13 The main driver for the incongruous results was the way in which the mutations were counted; the GFM study did not include missense mutations. In a subsequent collaboration between the two groups, a combined database confirmed the unfavorable prognostic values of ASXL1 nonsense and frameshift mutations when added to the clinical variables.14
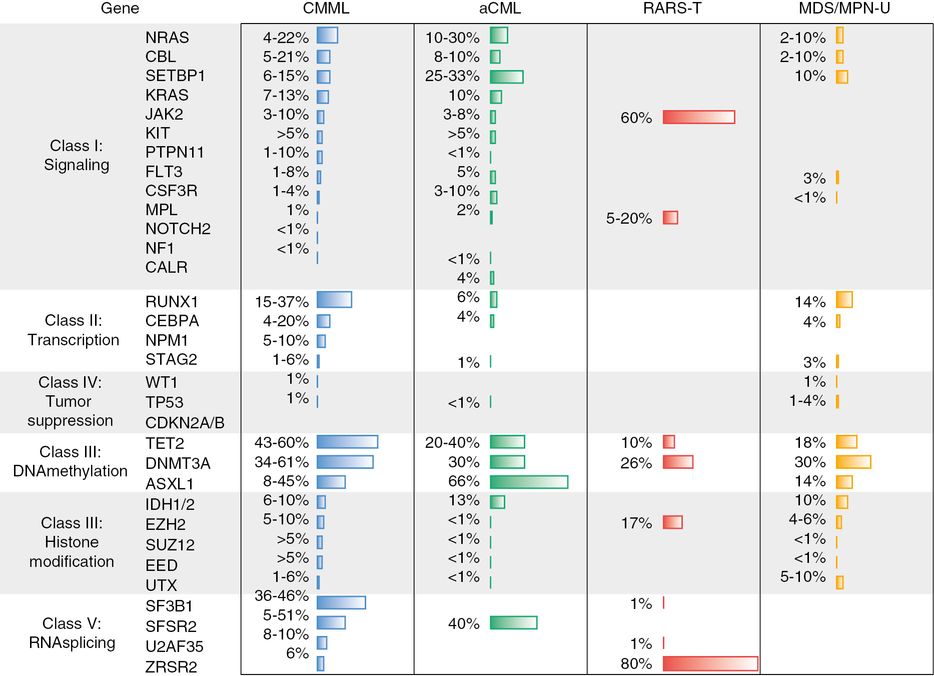
Frequency of mutation in myelodysplastic syndrome/myeloproliferative neoplasm overlap. CMML, chronic myelomonocytic leukemia; aCML, atypical chronic myeloid leukemia; RARS-T, refractory anemia with ringed sideroblasts and thrombocythemia; MDS/MPN-U, MDS/MPN overlap unclassified.
Related posts:
 The newly diagnosed patient with polycythemia vera
The newly diagnosed patient with essential thrombocythemia
Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
Accurately assessing risk in your myeloproliferative neoplasm patient
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
The newly diagnosed patient with polycythemia vera
The newly diagnosed patient with essential thrombocythemia
Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
Accurately assessing risk in your myeloproliferative neoplasm patient
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


