1 is most favorable and 10 least favorable
Circle the one number that describes, during the past week, how much difficulty you have had with each of the following symptoms
a Question used with permission from the MD Anderson Cancer Center Brief Fatigue Inventory.
The development of MPN-specific PRO tools offered an additional benefit – the ability to chronologically trend symptom severity and develop response criteria to therapies. The MPN-10 provided an optimal platform via which individual symptoms could be compared both to each other and to themselves across a continuum. The manipulation of the MPN-10 into quartiles as potential thresholds to assess symptomatic response was successfully demonstrated in a study using prospectively gathered MPN-SAF TSS data.7 MPN-10 scores of 0–7 (quartile 1), 8–17 (quartile 2), 18–31 (quartile 3), and ≥32 (quartile 4) statistically correlated with worsening symptom burden as clusters advanced (Table 6.2). Incorporation of the MPN-10 with quartile thresholds into routine evaluation of MPN patients provides a variety of benefits. First, assessment of symptom burden at the time of diagnosis assists in proactively uncovering patient symptomatology that might not otherwise be voluntarily disclosed. Furthermore, use of the tool during the disease course allows for direct comparison between individual symptoms over time to determine if therapies are effective, burdensome, or if there is evidence of disease progression.
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | |
|---|---|---|---|---|
| MPN-10 TSS | 0–8 | 9–18 | 19–32 | >32 |
| Essential thrombocythemia | 233 (50.1%) | 205 (45.9%) | 183 (39.5%) | 154 (32.3%) |
| Polycythemia vera | 161 (34.6%) | 152 (34%) | 169 (36.5%) | 172 (36.1%) |
| Myelofibrosis | 71 (15.3%) | 90 (20.1%) | 111 (24%) | 151 (31.7%) |
MPN-10 TSS, Myeloproliferative Neoplasm-10 Total Symptom Score.6
As information was collected on the ET symptom burden accrued, it became apparent that significant heterogeneity exists both within ET and in its association to other MPN subtypes. This includes wide variability in ET symptom prevalence (17–59%). In 2014, the MPN International Quality of Life Study Group aimed to further investigate this heterogeneity by assessing for the presence of clusters within each MPN disorder by using a unique heat-map cluster analysis of 1,470 MPN patients.8 The study additionally aimed to understand the relationship between these clusters and disease features and prognosis. This investigation identified the presence of five distinct ET clusters differing by MPN-10 score, age, gender, the presence of laboratory abnormalities, language and prior hemorrhage (all p < 0.05). Notably the clusters did not correlate with IPSET risk or the risk assessment variables of leukocytosis or history of thrombosis. Ongoing studies aim to uncover how the interrelationship between symptoms and disease features contributes to the development of MPN clusters.
Returning to the case, we apply the MPN-10, finding that the patient falls within symptom quartile 2, which represents a mild to moderate symptom burden. At this point, management with aspirin alone is deemed appropriate. The patient is followed again within a 2-year period. This timeline is reasonable given the patient’s relatively positive prognosis. During this visit, you find evidence of disease progression based on worsening fatigue, microvascular symptoms, and a rising platelet count. Reassessment with the MPN-10 has escalated the patient into symptom quartile 3, despite remaining low risk by IPSET criteria. Based on ELN guidelines, ET patients may be considered for cytoreductive therapy in the setting of a high-risk prognosis (age >60, major thrombotic or hemorrhagic event, increasing platelet count greater than 1,500 × 109/L), progressive myeloproliferation (increasing splenomegaly), or uncontrolled systemic symptoms. Considering this patient’s advancement to a higher symptom quartile, introduction of cytoreductive treatments would be reasonable. Once treatment has begun, monitoring of symptomatic response to therapy would be appropriate at 3–6-month intervals, or earlier should unexpected treatment effects be encountered. In the event that this patient’s symptoms advance despite front-line cytoreduction, consideration should be given to JAK2 inhibitor trials, interferon, or hydroxyurea therapy. Though still in early stages of investigation, cytogenetics are emerging as potential markers of disease severity and will likely play a greater role in prognostic algorithm designs. The CALR mutation is prevalent within the non-mutated JAK2 or MPL ET population and represents a positive prognostic marker with reduced incidence of thrombosis and leukemic transformation when present.9–11 The mutation additionally may support a positive response to therapies such as interferon-alpha.12 Though support has been waged to incorporate the CALR mutation into current prognostic scoring algorithms, it’s presence has not yet altered management guidelines.
Case 2
A 54-year-old woman is found to have a deep-vein thrombosis (DVT). Upon questioning, she has been having worsening problems with pruritus and fatigue. In her peripheral blood, it is found that she has a marked erythrocytosis with a hematocrit of 53%, a leukocyte count of 16 × 109/L, and a platelet count of 589 × 109/L. A bone marrow aspirate and biopsy are obtained and she has histologic features consistent with a WHO diagnosis of PV with normal cytogenetics, no increase in blasts, no reticulin fibrosis, and JAK2-mutated status with 60% allele burden. She is begun on therapy with hydroxyurea, phlebotomy, and aspirin. Additionally, an MPN-10 is administered for her at the initiation of her therapy and it is found that she has a symptom score of 37. The MPN-10 is monitored with her therapy, and her MPN-10 improves slightly, to a score of 24. On the maximally tolerated dose of hydroxyurea, she continues to require phlebotomy and display leukocytosis, and now she is having difficulties with mouth ulcerations. You feel that she is having an inadequate response despite being on a maximum tolerated dose of hydroxyurea and still has unmet symptomatic burden. You choose to initiate JAK2 inhibitor therapy as second-line therapy with ruxolitinib.
PV is characterized by erythrocytosis and, comparable to ET, an overall favorable prognosis for those patients who do not incur secondary complications. This patient’s initial evaluation demonstrates a number of key findings integral to determining the next step in treatment. To begin, we invoke the 2013 Leukemia PV Scoring System13 and determine that the presence of a prior vascular event (DVT) and a leukocyte count ≥15 × 109/L place her into the intermediate-risk prognostic category with a 15-year survival estimate. We additionally note that her MPN-10 symptom score integrates her into the highly symptomatic symptom quartile 4.
Management of PV is based on the ELN guidelines, whose treatment strategies were derived from the French Polycythemia Study Group Trials, the Polycythemia Vera Study Group, the European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study, the Cytoreductive Therapy in PV (Cyto-PV) trial, and the EORTC.1,14,15 As observed in this case, all PV patients, regardless of risk score, should be initiated on low-dose aspirin and undergo phlebotomy to keep hematocrit under 45%. By ELN guidelines, it would be appropriate to start cytoreduction in this patient just based on her symptomatology alone; she further qualifies for this treatment based on her history of DVT.
The patient’s MPN-10 score was appropriately followed after initiating therapy. Though improvements were noted in her total MPN-10 score, her symptom quartile3 score reflects she has only modestly improved with treatment. This finding is of relevance as the patient objectively demonstrates a statistically limited symptomatic response to current therapy. We are again faced with a number of pertinent questions. What role does symptom severity play in determining “therapy failure”? Are there therapies that specifically address PV symptoms? Can these therapies be used as adjuncts to conventional treatments?
To answer these questions, we must consider the evidence accrued on PV symptoms thus far. As was first demonstrated in the 2007 MPN internet symptom survey, PV patients suffer from significant symptom burden (fatigue: 84.9%, itching: 65.4%, night sweats: 49.1%, bone pain: 43.0%, fevers: 13.1%, weight loss: 9.6%), even in the setting of conventional therapies (aspirin: 72%, hydroxyurea: 53%, anagrelide: 22%, interferon: 16%, corticosteroids: 3%, other: 7%). It is well established that MPN therapies may worsen symptomatology by direct cytotoxic effects, induction of cytopenias, or leukemogenic transformation. Research has supported the notion that symptoms, such as pruritus, may play a positive or negative role in PV prognosis and, in 2011, the ELN subsequently included unmanaged symptoms as a PV response criteria marker.16
Until recently, PV patients exhibiting poor tolerance to conventional treatments had few alternative options. Detection of the JAK2 V617F mutation present in 96% of PV patients was a landmark advancement in the MPN field and propagated the development of numerous gene-targeted therapies, including JAK2 inhibitors, MTOR inhibitors, HDAC inhibitors, and antifibrosing agents. In 2012, the COMFORT trials highlighted the efficacy of the JAK2 inhibitor, ruxolitinib, in managing both the symptomatology and splenomegaly associated with MF.17,18
Ruxolitinib has recently been approved for treatment in patients with PV who are intolerant or resistant to hydroxyurea. In an evaluation of 34 PV patients receiving ruxolitinib for a median of 152 weeks, hematocrit levels <45 were achieved in 97% of recipients, with 44% demonstrating non-palpable spleens by week 24.19 Complete response criteria, as defined by modified 2009 ELN criteria, were achieved in 59% of participants. Responses were durable, with an 85% probability of maintaining a hematocrit <45% without phlebotomy for 48 weeks. Substantial reductions in leukocyte counts were achieved in 76% of recipients with baseline leukocytosis. Additionally, dramatic improvements were seen in symptoms (pruritus, night sweats, and bone pain) within 4 weeks of initiating therapy and were sustainable through week 144.
The RESPONSE trial, a randomized, multicenter open-label phase III investigation, aimed to evaluate ruxolitinib against best-available therapy in PV patients demonstrating resistance or intolerance to hydroxyurea, frequent phlebotomy requirements, inadequate hematocrit management, or splenomegaly (NCT01243944). Ruxolitinib proved effective in reducing spleen size and inducing complete hematological remission from baseline in 71.8% and 33% of treated patients vs. 23.6% and 8.9% of those receiving best-available therapy.20
As with ET, significant heterogeneity has been observed within the PV population. This heterogeneity was further investigated in the MPN Symptom Cluster study,8 where five unique PV clusters that differed by MPN-10, language, gender, spleen size, leukopenia, and laboratory abnormalities (all p<0.05) were uncovered. Total symptom score increased across each cluster as cluster size advanced. Specifically, no association was noted between symptom cluster and prognostic risk score, including the risk-scoring components of age and leukocytosis. The observance of symptom heterogeneity and individualized responses to therapies within PV has made the concept of using small-molecule inhibitors in combination an attractive model.
As we return to our case, we apply the ELN clinicohematologic response criteria and find that the patient’s leukocytosis, continued phlebotomy requirements, and unmanaged symptoms place her in the “no response” category to hydroxyurea therapy. As such, she is appropriately considered for JAK2 inhibitor therapy.
Case 3
A 69-year-old individual is found to have primary MF on the basis of worsening fatigue, splenomegaly, leukocytosis, and mild anemia. Laboratory studies demonstrate a hemoglobin of 9.8 g/dL, a leukocyte count of 26 × 109/L, a platelet count of 400 × 109/L, as well as 1–2% myelocytes and metamyelocytes in the peripheral blood with an absense of blasts. The bone marrow has 3+ reticulin fibrosis. The cytogenetics indicate the deletion of 20q–. On exam, the patient has a spleen which is palpable 18 cm below the left costal margin in the mid-clavicular line. He has an MPN-10 score of 46 out of 100 at the time of diagnosis.
The patient was initiated on front-line therapy with ruxolitinib at 10 mg twice a day. After 12 weeks of therapy, he described some improvement in symptomatic burden with a decreased MPN-10 score of 34. The spleen is reduced to 10 cm below the left costal margin. The platelet count improved to 250 × 109/L. On the basis of the partial benefit and the tolerability at the current dose, you decide to increase the dose of ruxolitinib to 15 mg twice daily and monitor the MPN-10. At this higher dose, the spleen reduces further to achieve an International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) clinical response. The MPN-10 now decreases to 18 with a 50% reduction from baseline and his blood counts remain stable.
MF carries the most clinically severe symptom burden and dismal disease course among the MPNs.21,22 This patient fits both the clinical characteristics and diagnostic criteria for MF based on the presence of constitutional symptoms, bone marrow reticulin and collagen fibrosis, peripheral leukoerythroblastosis, and splenomegaly,1 with the absence of molecular rearrangements suggestive of chronic myeloid leukemia. Our patient carries a characteristically “favorable” cytogenetics with the presence of a del(20q) mutation. Alternatively, normal karyotype or del(13q) also conveys a favorable prognosis in MF.23 Utilizing the Dynamic International Prognostic Scoring System (DIPSS),24 we find this patient falls into the high-risk scoring prognosis, with an age greater than 65 years, white blood cell count greater than 25 × 109/L, presence of constitutional symptoms, and a hemoglobin less than 10 g/dL. Many of this patient’s key clinical manifestations of MF are likely those which are primarily contributing to his severe symptom profile, namely the presence of splenomegaly leading to abdominal discomfort and early satiety, cytopenias leading to fatigue and inactivity, and inflammatory cytokine deregulation leading to an overall catabolic state with night sweats, fevers, and weight loss. We also note that this patient’s symptom severity falls within the highest symptom quartile scoring (MPN-10 score >32).
Treatment of MF is clinically oriented due to the lack of therapies capable of offering longstanding disease remission, with the notable exception of allogeneic stem cell transplantation. Many suggested management guidelines exist to assist in the treatment of MF patients, including guidelines put forth by ELN,1 as well as individual treatment algorithms such as those developed by Tefferi25 and Vannucchi.26 These treatment algorithms emphasized the importance of considering the benefits and risks of cytoreduction and spleen reduction, the availability of novel or targeted treatments, performance status, and patient wishes, but did not incorporate an objective assessment of symptom burden. This was not surprising given that these treatment algorithms were developed during an era where traditional therapies were able to reduce splenomegaly and alleviate cytopenias, but did not result in significant symptom or mortality benefit and came at the cost of substantial negative side effects. These primarily palliative treatments included hydroxyurea, cladribine, melphalan/busulfan, radiation, and splenectomy. The past few years have ushered in the development of novel targeted treatments such as mammalian target of rapamycin inhibitors,27 selective and non-selective Janus kinase inhibitors,17,28,29 and thalidomide analogs.30 Despite not appearing to have specific anticlonal activity, these novel agents have displayed excellent progress in reducing spleen size, reducing cytopenias, and improving symptom burden. Given this, assessment of symptom response has become a vital component to monitoring MF treatment and therapeutic response and a key factor in the decision of what therapeutic agent may lead to the greatest improvement in quality of life. However, this leads us to multiple key queries in this patient’s care. Which patients would most benefit from novel therapies? Which of the novel therapies should be chosen to treat this patient’s symptom burden, splenomegaly, and cytopenias?
Recent trials of novel treatments for MF have found promising results. Ruxolitinib is the first medication approved by the US FDA for the initial treatment of MF patients with intermediate-2 or high-risk MF. Ruxolitinib, a JAK2 inhibitor, is the most robustly studied of the novel treatments. It is a non-selective inhibitor that is effective in decreasing cytokine activation and hematopoietic proliferation.31,32 In two key randomized, double-blind studies, ruxolitinib induced substantial symptom burden reduction and alleviation of spleen size when compared against placebo (COMFORT-I) and best-available therapy (COMFORT-II).17,18 Two additional JAK2 inhibitors, momelotinib (CYT387) and pacritinib (SB1518), have been shown to be efficacious in reducing splenomegaly, MPN-related symptom burden, and cytopenias.28,33 Thalidomide, lenalidomide, and pomalidomide are immunomodulatory agents with success in reducing anemia and thrombocytopenia.30,34,35 MTOR inhibitors such as everolimus and histone deacetylase inhibitors such as givinostat and panobinostat cause reduced splenomegaly and offer some relief of symptom burden.36–38
Prior to determining the best clinical treatment strategy, a clinician must first evaluate prognosis based on established prognostic scoring algorithms and objectively assess symptom burden (Figure 6.1 B). Prior to therapy initiation, symptom burden should be individually assessed by MPN risk score (IPSS/DIPSS criteria) and symptoms (MPN-10). Symptomatic groupings can be obtained by fractionating MPN-10 symptom burden into scores based on quartile ranking (MPN-10 quartile 1: <8; MPN-10 quartile 2: 8–17; MPN-10 quartile 3: 18–31; MPN-10 quartile 4: ≥32). For individuals with low-risk disease, non-severe symptom burden, and small spleen size, observation or interferon treatment may be considered. Alternatively, individuals with high symptom burden (MPN-10 score ≥18, indicating third or fourth symptom quartile status) should be considered for JAK2 inhibitor treatment given the dramatic symptomatic and splenic responses observed with JAK2 inhibitor therapy. Although ruxolitinib was chosen in this case, there are numerous alternative JAK2 inhibitors currently undergoing clinical trials that may also convey a desirable symptom response. Dosing on these agents should be uptitrated as tolerable, keeping in mind the patient’s cytopenias, which can also be modified using steroids, androgens, immunomodulatory agents, or erythropoiesis-stimulating agents. Patients with unfavorable mutational status should be considered for autologous stem cell transplant.25 Although autologous stem cell transplant may be potentially curative, it is complicated by a high overall 1-year treatment-related mortality of 30% and overall mortality of 50%39 and is only useful in a select subset of patients.1 Traditional treatments such as hydroxyurea, cladribine, oral melphalan, or busulfan can also be considered in patients who are intolerant or resistant to ruxolitinib, but are likely of limited clinical usefulness. Only individuals refractory to traditional and experimental therapies should be considered for splenectomy or radiotherapy. If MF progresses to AML, pharmacological interventions are of limited utility and median survival is diminished to 2.6–5 months.40
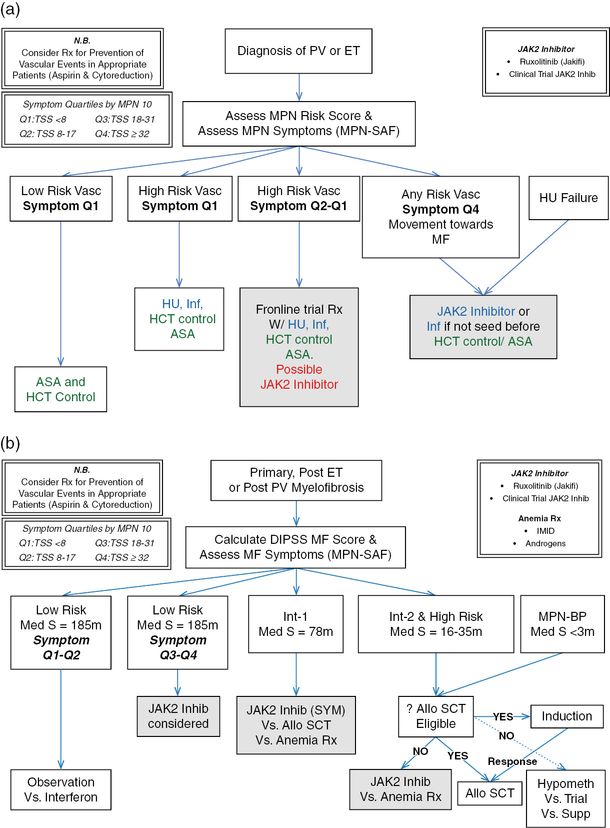
(a) Management strategies for essential thrombocythemia (ET) and polycythemia vera (PV).
(b) Management strategies for myelofibrosis (MF). MPN, myeloproliferative neoplasm; MPN-SAF, Myeloproliferative Neoplasm Symptom Assessment Form; HU, hydroxyurea; ASA, aspirin; HCT, hematocrit; Inf, interferon; Rx, prescription; DIPSS, Dynamic International Prognostic Scoring System; IMID, immunomodulatory derivative of thalidomide; MPN-BP, MPN blast phase; allo SCT, allogeneic stem cell transplantation; Q1, first symptom quartile; Q2, second symptom quartile; Q4, fourth symptom quartile; Med S, median survival; M, months.
Related posts:
 Hypereosinophilia: an illustrated approach to diagnosis and management
Familial myeloproliferative neoplasms – implications for patients and their families
Accurately assessing risk in your myeloproliferative neoplasm patient
Myelodysplastic/myeloproliferative neoplasm overlap syndromes
Systemic mast cell disease: diagnosis and management
Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Hypereosinophilia: an illustrated approach to diagnosis and management
Familial myeloproliferative neoplasms – implications for patients and their families
Accurately assessing risk in your myeloproliferative neoplasm patient
Myelodysplastic/myeloproliferative neoplasm overlap syndromes
Systemic mast cell disease: diagnosis and management
Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


