Peripheral blood smear in patient 1 shows dysplastic granulocytes, with hypogranular cytoplasm and megaloblastoid chromatin. Two myelocytes (top) are part of a significant left shift. A segmented neutrophil (lower right) contains a bi-lobed nucleus, another dysplastic feature.
Bone marrow core biopsy showed a markedly hypercellular marrow, at approximately 95% average cellularity (Figure 15.2). Trilineage hematopoiesis was present. Megakaryocytes were present at a normal frequency, and while most megakaryocytes appeared normal, a significant minority (around 20%) was dysplastic (Figure 15.3). Immunohistochemistry was negative for increased blasts or increased plasma cells. A reticulin stain showed diffuse mild to moderate fibrosis of the marrow space.
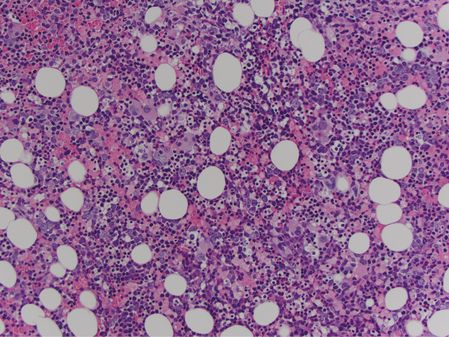
Marrow core biopsy in patient 1 shows a markedly hypercellular marrow.
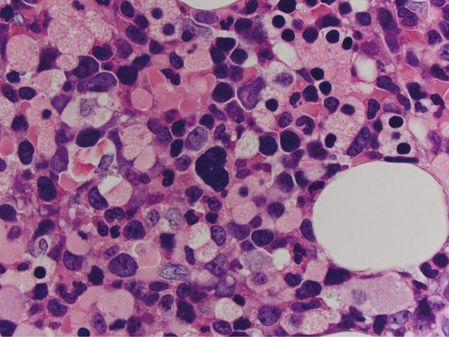
A dysplastic megakaryocyte (center) in patient 1’s marrow core biopsy possesses a hyperchromatic, poorly lobated nucleus.
Flow cytometry of the aspirate was negative for increased blasts, revealed 5% monocytes, and showed granulocytic abnormalities compatible with dysgranulopoiesis. Chromosomal analysis identified an abnormal clone, accounting for all 20 analyzed cells, containing gain of one copy of chromosome 8 (trisomy 8) as its sole abnormality. Fluorescent in situ hybridization (FISH) assays confirmed trisomy 8, and were negative for other abnormalities, including BCR-ABL1. Molecular analysis of the blood was negative for the JAK2 V617F mutation.
The combination of proliferative (prominent neutrophilia) and dysplastic (anemia, thrombocytopenia, dysplastic morphology in the megakaryocytic and neutrophilic lineages) features, in the absence of criteria diagnostic of acute myeloid leukemia (AML), suggests a condition in the WHO category of myelodysplastic syndrome/MPN (MDS/MPN). In an adult, this leads to a consideration of two diagnoses: aCML and chronic myelomonocytic leukemia (CMMoL). Although this patient had an absolute monocytosis, it was minimal.
In CMMoL, monocytes are absolutely and relatively increased, and almost always account for 10% or more of peripheral WBCs (the absolute monocyte count is frequently mildly increased in aCML, but in the absence of a significant relative increase). While significant neutrophilia, which can appear dysplastic, occurs in roughly half of CMMoL cases, this entity typically lacks a significant left shift, with immature granulocytes accounting for <10% of peripheral neutrophilic granulocytes. In contrast, in aCML immature granulocytes typically account for more than 10% of peripheral neutrophilic granulocytes. In this case, the cytogenetic findings do not help differentiate aCML from CMMoL, and a diagnosis of aCML was made.
Patient 2 was a 57-year-old male noted on routine blood work to have a WBC of 25,000/μL. Initial workup failed to reveal a secondary cause of the leukocytosis. A repeat complete blood count 2 months later revealed the leukocytosis had worsened to 44,000/μL. The patient was referred to a hematologist, and the leukocytosis had progressed to 92,000/μL, with a manual differential showing 86% segmented neutrophils, 5% bands, 1% metamyelocytes, 1% myelocytes, 0% blasts, 6% lymphocytes, and 0% monocytes, eosinophils, and basophils. There was thrombocytopenia, with a platelet count of 72,000/μL. The hemoglobin was mildly decreased, at 12.3 g/dL, and the MCV was within normal limits, at 96 fL.
Review of a stained blood smear confirmed the presence of a marked neutrophilia, composed predominantly of mature segmented neutrophils, and with only a slight left shift. The neutrophils appeared normal morphologically, without dysplastic features, and were negative for toxic changes. Basophils and blasts were extremely rare. Molecular assays of peripheral blood were negative for BCR-ABL1 fusion products by reverse transcriptase polymerase chain reaction (RT-PCR), and negative for JAK2 V617F mutation by PCR. Flow cytometry of peripheral blood confirmed a predominance of granulocytes, which appeared normal, and showed only rare circulating myeloblasts.
A bone marrow core biopsy was markedly hypercellular, at nearly 100% cellularity (Figure 15.4), with the large majority found to be neutrophilic granulocytes. Megakaryocytes were present at a slightly decreased frequency relative to overall marrow cellularity. The megakaryocytes appeared normal, possessing neither significant atypia nor dysplastic features, and were not abnormally clustered (Figure 15.5). Immunohistochemistry of the core was negative for increased blasts or increased plasma cells. In situ hybridization studies showed that the few plasma cells present showed a normal ratio of kappa to lambda light-chain expression. A reticulin stain was negative for significant reticulin fibrosis.
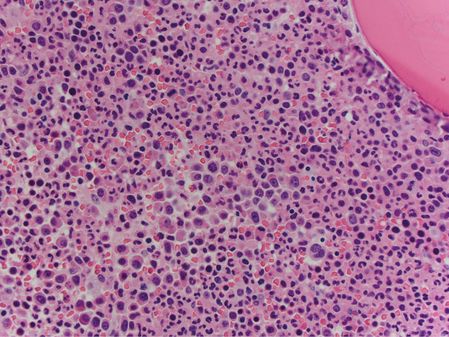
Marrow core biopsy from patient 2 is nearly 100% cellular, with a marked predominance of granulopoietic elements.
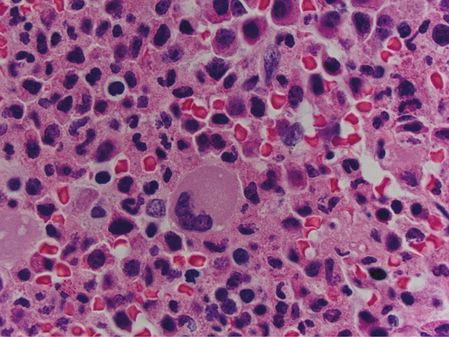
In the marrow core biopsy from patient 2, megakaryocytes are not clustered, and are morphologically within normal limits.
The marrow aspirate smears confirmed the increased myeloid:erythroid ratio, the absence of increased blasts, and the absence of significant morphologic evidence of dyshematopoiesis. Cytogenetic analysis of the aspirate showed a normal male karyotype, and was negative for the BCR-ABL1 gene fusion by FISH.
The main differential diagnoses included a marked reactive neutrophilia (so-called leukemoid reaction) vs. a hematopoietic neoplasm. The persistent worsening of the neutrophilia in the absence of any identifiable infectious or inflammatory condition, the lack of toxic changes in the neutrophils, and the lack of a significant left shift of the neutrophils all argued against a reactive neutrophilia. Among hematopoietic neoplasms, the main considerations were MPN, MDS/MPN overlap condition, and the well-described phenomenon of a plasma cell neoplasm with an associated marked neutrophilia. The absence of increased plasma cells in the marrow and the polytypic appearance of these plasma cells on special studies excluded the latter. Given the persistent thrombocytopenia and mild anemia, MDS/MPN was considered. However, there were insufficient dysplasia-related findings to allow for this diagnosis. Specifically, there was no significant morphologic evidence of dysplasia, cytogenetics did not reveal an MDS-related abnormality, and blasts were not increased and were phenotypically normal. The absence of monocytosis excluded CMMoL.
The patient was therefore diagnosed with an MPN. The absence of the BCR-ABL1 fusion by multiple testing modalities definitively excluded CML. The thrombocytopenia excluded essential thrombocythemia, and would have been unusual for polycythemia vera (PV) or for the prefibrotic stage of primary myelofibrosis (PMF). The absence of a JAK2 mutation essentially excluded PV, and made PMF less likely. Finally, the lack of megakaryocytic clustering and megakaryocytic atypia excluded PMF. The patient was therefore diagnosed with CNL, due to prominent leukocytosis, with neutrophils and bands comprising over 80% of the leukocytes, the absence of a significant left shift, no alternate explanation for the neutrophilia, and exclusion of criteria for any other MPN.
Background
aCML is a clonal stem cell disorder, classified by the WHO as an MDS/MPN overlap condition,1 and is characterized by the presence of leukocytosis made up of a predominance of dysplastic neutrophils. Although it shares many morphologic and clinical features with CML, it lacks the BCR-ABL1 fusion gene. This is a rare disease, with only 1–2 diagnoses for every 100 cases of CML.2 Patients are typically older at presentation, with a median age between 60 and 70 years, and the male-to-female ratio is roughly even.3–5
CNL is classified by the WHO as an MPN characterized by neutrophilia in the peripheral blood, and similar to aCML, by the absence of a BCR-ABL1 fusion gene. It is a diagnosis of exclusion when other considerations that could account for neutrophilia have been investigated and ruled out.1 This is an extremely rare entity, with only about 200 known reported cases.1 Although the etiology is unknown, a clonal cell assay study suggested it originates from granulocyte-committed precursors.6 Like aCML, most patients are diagnosed later in life, and there is also no known gender bias for its incidence.
Diagnosis
A minimum threshold of a WBC count ≥ 13,000/μL has been adopted for diagnostic purposes in aCML,1,7 but many patients have very high counts. Most have anemia and thrombocytopenia, and many have hepatosplenomegaly.8 The bone marrow is typically hypercellular, and reticulin fibrosis can be seen. Morphologically, leukemic blasts are rarely increased but neutrophilic precursors such as myelocytes and metamyelocytes are common. Monocytes account for <10% of cells, and although basophils can be elevated, they are rarely as high as observed in many cases of CML. Dysgranulopoiesis and dyserthryropoeisis are common features of aCML. Patients lack BCR-ABL1 as well as rearrangements of PDGFRA and PDGFRB, and in the majority of cases have a normal karyotype. When they do occur, the most common cytogenetic abnormalities are trisomy 8 and del(20)q.9,10
CNL patients present with neutrophilia; the minimum WBC for diagnosis is at least 25,000/μL.1 Immature forms including blasts are not present, nor are increased monocytes or basophils. Patients may have symptoms of bone marrow failure, gout, or pruritus at diagnosis. The bone marrow is hypercellular with a predominance of neutrophilic granulocytes, and occasionally, proliferation of erythroid forms and megakaryocytes.11,12 While any tissues can be infiltrated, hepatosplenomegaly is expected.1 In contrast to aCML, there is no evidence of reticulin fibrosis or dysplasia in any lineage. Similarly to aCML, the majority of patients have a normal karyotype, but clonal evolution involving the development of new chromosomal abnormalities associated with clinical progression can occur.13
As illustrated by patient 2 in the case description, CNL is a diagnosis of exclusion, and the lack of a BCR-ABL1 translocation is the first criterion that must be met. Additionally, there cannot be evidence of rearrangements of FGFR1, PDGFRA, or PDGFRB. Patients with the variant BCR-ABL1 break point between c3 and c4 of BCR have a 230 kD fusion protein which can result in predominant neutrophilia;14 however, this entity, as well as other patients with cryptic BCR-ABL1 rearrangements, should be classified as CML, not CNL.15 Neutrophilia can be reactive, due to infection, inflammation, or associated with an underlying malignancy; it is commonly observed with an underlying plasma cell dyscrasia, which, along with other reactive causes, needs to be ruled out to diagnose CNL.1
Table 15.1 gives an overview of the WHO criteria for diagnosis of CNL and aCML, and Figure 15.6 presents a diagnostic algorithm for these conditions.
| Chronic neutrophilic leukemia | Atypical chronic myeloid leukemia | |
|---|---|---|
| Peripheral blood |
|
|
| Bone marrow |
|
|
| Organs |
| – |
| Exclusions |
|
|
WBC, white blood cells; PV, polycythemia vera; MF, myelofibrosis; ET, essential thrombocythemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm.
Related posts:
 Which patients are candidates for JAK inhibitors and how to manage patients on JAK inhibitor treatment
Hypereosinophilia: an illustrated approach to diagnosis and management
Familial myeloproliferative neoplasms – implications for patients and their families
Hematopoietic cell transplantation for myelofibrosis: who and when?
Myelodysplastic/myeloproliferative neoplasm overlap syndromes
Systemic mast cell disease: diagnosis and management
Which patients are candidates for JAK inhibitors and how to manage patients on JAK inhibitor treatment
Hypereosinophilia: an illustrated approach to diagnosis and management
Familial myeloproliferative neoplasms – implications for patients and their families
Hematopoietic cell transplantation for myelofibrosis: who and when?
Myelodysplastic/myeloproliferative neoplasm overlap syndromes
Systemic mast cell disease: diagnosis and management
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


