Megakaryocytes
Amy E. Geddis
Megakaryocytes are large polyploid cells within the bone marrow that produce circulating platelets. Platelets are small anucleate cells that play an essential role in hemostasis, both serving to plug holes in blood vessels and initiate coagulation. To function in this role, platelets have unique properties, including the ability to adhere to injured blood vessels and to other platelets, undergo dramatic shape change, release granules containing vasoactive and thrombogenic substances, and modulate the phospholipid content of their external membrane. Platelets also release agents that promote wound healing. Based on the adult blood volume (5 L), the number of platelets per microliter of blood (˜2 × 105), and their circulatory half-life (10 days), it can be calculated that each day an adult human produces 1 × 1011 platelets, although in times of increased demand, platelet production can rise tenfold or more. The process of megakaryocyte and platelet formation is complex and only partially understood. This chapter discusses the molecular mechanisms underlying the development of megakaryocytes and platelets, as well as disease states that have illuminated our understanding of megakaryopoiesis.
HISTORICAL PERSPECTIVE
Platelets were described by Addison in 1841 as “extremely minute … granules” in clotting blood1 and were termed platelets (blutplättchen) by Bizzozero, who also observed their adhesive qualities as “increased stickiness when a vascular wall is damaged.”2 The same elements were identified by microscopic examination of blood smears by Osler and Schaefer and by Hayem in the late nineteenth century.3,4 Duke, a student of James Homer Wright’s, associated low platelet counts, or thrombocytopenia, with clinical bleeding that could be corrected by transfusion.5
Megakaryocytes are rare polyploid cells within the bone marrow, representing approximately 0.05% of nucleated cells. Although Howell coined the term megakaryocyte as early as 1890, it was not until 1906 that James Homer Wright put forth the hypothesis that blood platelets are derived from the cytoplasm of megakaryocytes,6,7 and the basic elements of thrombopoiesis were established. Many years later in the early 1990s, c-Mpl, the cellular homolog of the v-Mpl (myeloproliferative leukemia retrovirus) oncogene, was identified and proposed to be a hematopoietic growth factor receptor.8,9,10 and 11 Shortly thereafter, thrombopoietin (TPO) was cloned based on its properties of binding to and stimulating growth of c-Mpl-bearing cells; subsequent characterization of the cytokine revealed it to be the major growth factor promoting megakaryopoiesis.12,13,14,15 The availability of TPO was a major step forward in megakaryocyte and platelet biology as it facilitated expansion of megakaryocytes for further study as well as dissection of the signaling pathways regulating megakaryocyte proliferation and survival.
This chapter focuses primarily on mammalian species. However, it is worth noting that in nonmammalian vertebrates, such as fish and birds, the hemostatic equivalent of platelets is served by nucleated thrombocytes,16 and polyploid megakaryocytes are not present. The purpose of megakaryocyte polyploidization in the process of platelet development is not understood but it is speculated that this allows for increased expression of the proteins that are needed for the production of large numbers of platelets.17
MEGAKARYOPOIESIS AND THE HEMATOPOIETIC STEM CELL
Megakaryocytes, like cells of all blood lineages, are derived from the hematopoietic stem cell (HSC). HSCs are also very rare cells within the bone marrow, estimated to represent approximately 0.01% of total nucleated cells. Identified in 1961 by Till and McCullough,18 key properties of HSCs include the capacity for prolonged quiescence, self-renewal, and ultimately expansion and differentiation into the total diversity of blood cell types. Although megakaryocytes are terminally differentiated cells, they share a number of characteristics with HSCs.19 For example, both megakaryocytes and HSCs express the integrins CD41 (integrin α2b) and CD61 (integrin β3). The integrin α2bβ3 complex forms a fibrinogen receptor that is one of the defining markers of the megakaryocyte lineage, but is now known to be expressed much earlier in hematopoiesis, including on HSCs.20,21 and 22 In addition, expression of integrin β3 in association with integrin αν identifies a population of cells with enhanced long-term repopulating activity in transplantation assays.22 Several transcription factors share key roles in both HSC and megakaryocyte development, including SCL/TAL1, RUNX1, GATA, and ETS family members (discussed below). Strikingly, even the global transcriptional profiles of HSCs and early megakaryocytic/erythroid progenitors (MEPs) are highly similar.23
In addition to similarities in cell surface protein and gene expression patterns, both HSCs and megakaryocytes require TPO for their growth and survival. The importance of TPO and its receptor c-Mpl for the function of HSCs was suspected when knockout mice, lacking either TPO or c-Mpl, were noted to be deficient in progenitors of all hematopoietic lineages. Transplantation experiments in mice confirmed that in the absence of an intact TPO-c-Mpl axis, HSCs are unable to compete with wild-type cells, either due to reduced numbers or expansion potential, although the mice do not develop bone marrow failure.24,25,26,27,28 In humans, mutations that eliminate c-Mpl function are the basis of congenital amegakaryocytic thrombocytopenia (CAMT). Children with CAMT are born with isolated thrombocytopenia and a paucity of megakaryocytes, but typically progress to trilineage bone marrow failure within the first decade of life, providing evidence of an essential and nonredundant role of TPO for the HSC.29
TPO has been demonstrated to contribute to both HSC quiescence and expansion in various experimental settings. In mice, the absence of TPO or c-Mpl leads to acceleration of HSC cell cycling and eventual HSC exhaustion, suggesting that TPO is required for HSC quiescence and self-renewal.30 One potential mechanism
by which TPO enhances HSC quiescence is by up-regulation of cyclin-dependent kinase inhibitors.30,31 In addition, TPO affects the interactions between the HSC and the bone marrow niche. HSCs are maintained in a quiescent state through interactions with TPO-producing osteoblasts.31 Although both outside-in and inside-out signaling, TPO triggers a conformational change of integrin ανβ3 to its high-affinity state, and binding of ανβ3 to its extracellular ligand within the niche results in phosphorylation of tyrosine 747 (Y747) of the integrin β3 tail, which cooperates with TPO signals to inhibit cell cycling and maintain HSC long-term repopulating activity.32
by which TPO enhances HSC quiescence is by up-regulation of cyclin-dependent kinase inhibitors.30,31 In addition, TPO affects the interactions between the HSC and the bone marrow niche. HSCs are maintained in a quiescent state through interactions with TPO-producing osteoblasts.31 Although both outside-in and inside-out signaling, TPO triggers a conformational change of integrin ανβ3 to its high-affinity state, and binding of ανβ3 to its extracellular ligand within the niche results in phosphorylation of tyrosine 747 (Y747) of the integrin β3 tail, which cooperates with TPO signals to inhibit cell cycling and maintain HSC long-term repopulating activity.32
TPO can also promote HSC expansion. In vitro, single cell experiments demonstrated that TPO supports the survival of HSCs, and in addition promotes their proliferation if either stem cell factor (SCF) or interleukin (IL)-3 is also present.33 TPO also cooperates with vascular endothelial growth factor (VEGF) through an autocrine mechanism; TPO increases VEGF expression in primitive murine hematopoietic cells through induction of hypoxia-inducible factor (HIF)-1α, and TPO in conjunction with VEGF promotes proliferation of hematopoietic progenitors.34 In human embryonic stem cell models, the combination of TPO and VEGF enhances the generation of CD34+ cells.35 In addition to HIF-1α, TPO increases the expression and function of HOX family members. TPO-induced p38 MAPK enhances HOXB4 expression, a homeobox factor that promotes HSC expansion.36,37 TPO-induced MAPK also leads to phosphorylation of MEIS1 which binds to and facilitates nuclear translocation of HOXA9.38 As with HOXB4, disruption of HOXA9 in animal models also leads to HSC defects.39 Finally, Lnk, an adaptor protein that modulates TPO-induced STAT5, AKT, and p38 MAPK signaling, negatively regulates self-renewal of HSCs. Lnk-deficient HSCs are hypersensitive to TPO and have enhanced long-term repopulating activity due to increased self-renewal.40 Although the mechanisms remain incompletely understood, clinical studies are examining the possible applications for TPO-receptor agonists in expanding HSCs in patients with acquired aplastic anemia with promising preliminary results.41
MEGAKARYOCYTE DEVELOPMENT
In current models of hematopoiesis, the HSC can either undergo symmetric divisions, resulting in self-renewal, or asymmetric divisions, in which the daughter cells become progressively committed to a specific hematopoietic lineage. Lineage commitment is marked by characteristic gene expression patterns and modulated by hematopoietic cytokines (Fig. 15.1). Specific multipotent and megakaryocytic progenitors can be defined using semi-solid colony assays. More recent strategies have used fluorescence-activated cell sorting approaches to identify progenitors and committed cells prospectively based on cell surface protein expression patterns42,43 (Table 15.1). The earliest multipotent progenitor that has been isolated is the committed myeloid progenitor, which gives rise to the colony-forming-unitgranulocyte-erythroid-macrophage-megakaryocyte (CFU-GEMM). From the CFU-GEMM arises a bipotential progenitor with both megakaryocytic and erythroid potential, the MEP.44,45,46 and 47 Identified within the CD34+CD38low cell population of progenitor cells,46 murine MEPs have been further defined as existing within the CD150+CD9loendoglinlo fraction of Lin−cKit+IL7Rα−FcγRII/IIIloSca1− cells.48,49 Megakaryocyte-lineage-committed progenitors in contrast were included in the CD150+CD9hiendoglinlo fraction. The most primitive committed megakaryocytic cell is the megakaryocytic burst-forming unit, which resembles a small lymphocyte and forms a colony of 40 to 500 cells in semi-solid assays. The megakaryocytic colony-forming unit (CFU-MK) is a more mature progenitor that will form a colony of 3 to 50 cells in semi-solid assays. Whereas all megakaryocytic progenitors can be recognized by staining for CD41 and, in murine cells, acetylcholinesterase, only CFU-MK express HLA-DR. Though this represents the classical model of megakaryocytic differentiation, it has been proposed that an alternative pathway exists in which the MEP can arise directly from the HSC.50
Precursors that are committed to the megakaryocyte lineage undergo further maturation before they are capable of platelet production. As they differentiate, megakaryocytes express specific cell surface proteins, become polyploid, develop a complex cytoplasm containing granules and a system of demarcation membranes, and ultimately form proplatelet processes (Fig. 15.2). Historically, the developmental stages of megakaryocytes have been described based on morphologic criteria including the quality and quantity of the cytoplasm and the size, lobulation, and chromatin pattern of the nucleus. Thus, cells can be categorized as promegakaryoblasts and stage I megakaryoblasts, stage II promegakaryocytes, stage III mature megakaryocytes, and stage IV megakaryocytes preparing to release platelets from their cytoplasm. Stage I megakaryocytes account for approximately 25% of all megakaryocyte lineage cells in a normal marrow. They are 6 to 24 µm in diameter and contain a relatively large, minimally indented nucleus (2 to 4 N) with loosely organized chromatin and multiple nucleoli and scant basophilic cytoplasm containing a small Golgi complex, a few mitochondria and α-granules, and abundant free ribosomes. Although the cytoplasm is scant, the demarcation membrane system (DMS) that is characteristic of more mature cells has already begun to form. This early, proliferative megakaryocytic cell is also sometimes termed a megakaryoblast and, in rodent hematopoiesis, is characterized by intense staining for acetylcholinesterase. Stage II megakaryocytes also represent approximately 25% of marrow megakaryocytes. They measure 14 to 30 µm in diameter, with a lobulated nucleus of 8 to 64 N and more abundant polychromatic cytoplasm. Ultrastructurally, the cytoplasm contains more abundant α-granules and organelles, and the DMS begins to expand at this stage of development. The remaining 50% of morphologically recognizable megakaryocytes in the marrow are stage III/IV megakaryocytes. These cells are very large (40 to 60 µm in diameter) with abundant mature cytoplasm. The DMS is prominent, and the multilobulated nucleus becomes more compact and is often eccentrically placed. Stage IV megakaryocytes may display proplatelet processes. It has been calculated that it takes approximately 72 hours for stage III and stage IV cells to develop from stage I megakaryocytes.
Starting in early megakaryocytes, expression of characteristic cell surface proteins, including integrin α2bβ3 and GPIb/IX, becomes functionally important. Integrin α2bβ3 is an integral transmembrane protein that acts as a receptor for fibrinogen. Of the two subunits, only integrin α2b is megakaryocyte lineage specific. Loss of integrin α2bβ3 leads to Glanzmann thrombasthenia due to failure of the defective platelets to engage fibrinogen during aggregation. The two subunits of integrin α2bβ3 are synthesized in the endoplasmic reticulum and form a Ca2+-dependent complex immediately on translation, a step necessary for membrane expression. Subsequently, the α-subunit is cleaved into heavy and light chains and modified with carbohydrate before transfer to the cell surface, demarcation, and α-granule membranes.51,52 Megakaryocytes and platelets contain approximately twice the amount of integrin α2bβ3 in granules as is present on the cell surface, the granule compartment serving as a mobilizable pool that is exteriorized on platelet activation.
The GPIb/IX complex is developmentally expressed only slightly later than integrin α2bβ3 and forms a receptor for von Willebrand factor (vWF).21 Although known for its role in binding vWF, GPIbα also is an important structural component in platelets through its interaction with the cytoskeletal protein filamin A. Filamin A, in turn, promotes clustering and activation of GPIb, thus regulating its function in platelet aggregation.53,54 Absence of
the GPIb/IX complex results in Bernard-Soulier syndrome (BSS), an autosomal recessive macrothrombocytopenia with severe platelet dysfunction; mutations of filamin A are also associated with macrothrombocytopenia but GPIb surface expression is normal.55 Except for minor expression in endothelial cells,56 GPIb is a megakaryocyte-specific protein. GPV is also expressed in complex with GPIb and GPIX in a 2:2:1 (Ib:IX:V) stoichiometric ratio57,58; however, GPV is not required for the function of the GPIb/V/IX complex in binding vWF. Genetic elimination of GPV has little effect on platelet adhesion59 and no mutations of GPV have been associated with BSS.60 Rather, GPV appears to function as a target of thrombin, possibly playing a role in platelet activation.61,62
the GPIb/IX complex results in Bernard-Soulier syndrome (BSS), an autosomal recessive macrothrombocytopenia with severe platelet dysfunction; mutations of filamin A are also associated with macrothrombocytopenia but GPIb surface expression is normal.55 Except for minor expression in endothelial cells,56 GPIb is a megakaryocyte-specific protein. GPV is also expressed in complex with GPIb and GPIX in a 2:2:1 (Ib:IX:V) stoichiometric ratio57,58; however, GPV is not required for the function of the GPIb/V/IX complex in binding vWF. Genetic elimination of GPV has little effect on platelet adhesion59 and no mutations of GPV have been associated with BSS.60 Rather, GPV appears to function as a target of thrombin, possibly playing a role in platelet activation.61,62
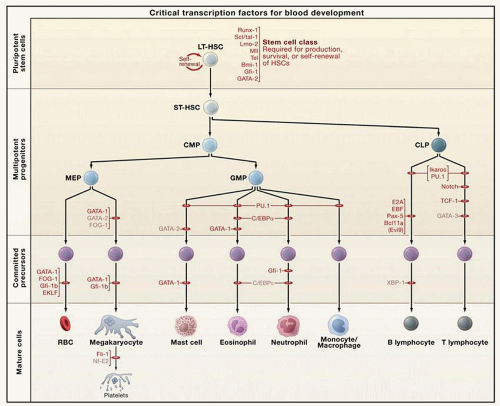 FIGURE 15.1. Hematopoietic cell development. Transcription factors regulate the differentiation of the HSC to produce committed progenitors and mature blood cells. The MEP is a bipotential precursor that gives rise to both erythroid and megakaryocytic lineages. Whereas GATA1 and FOG1 are required for both erythropoiesis and megakaryopoiesis, EKLF favors red cell development and FLI1 promotes megakaryopoiesis. With permission from Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008;132:631-644. |
TABLE 15.1 CELL SURFACE MARKERS CHARACTERISTIC OF MEGAKARYOCYTE DEVELOPMENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
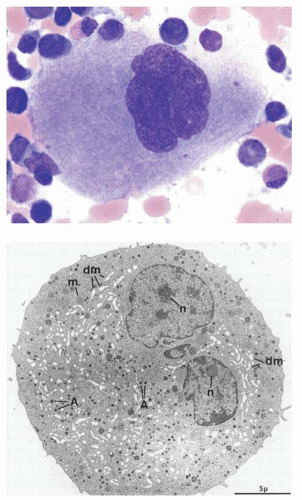 FIGURE 15.2. Mature megakaryocytes (A) Bone marrow megakaryocyte visualized by Wright’s stain. Note the multilobated nucleus. (B) Electron micrograph of a mature megakaryocyte, showing cytoplasmic alpha granules (A), mitochondria (m), and demarcation membranes (dm). There appear to be separate nuclei with nucleoli (n), but these represent two lobes of a single nucleus visible in this section. |
One of the most characteristic and intriguing features of megakaryocyte maturation is the development of polyploidy. As they mature, megakaryocytes switch from mitotic cell cycling to endomitosis, or DNA replication in the absence of nuclear or cytoplasmic division (Fig. 15.3). Endomitosis begins late in stage I megakaryoblasts, after sufficient diploid cell divisions have occurred to expand the number of megakaryocytic precursor cells, and is completed by the end of stage II megakaryocyte development.63,64 This carefully controlled process (i.e., cells are polyploid, rather than aneuploid) results in cells containing DNA content from 8 to 128 times the normal chromosomal complement in a single, highly lobated nucleus. Endomitosis is not simply the absence of mitosis, but rather an aborted mitosis.65 The cell-cycle kinetics of endomitotic cells is also unusual, characterized by a short G1 phase, a normal or modestly prolonged DNA synthesis phase, a short G2 phase, and a very short endomitosis phase.66 During the latter, megakaryocytic chromosomes condense, the nuclear membrane breaks down, and centromeres form mitotic spindles on which the replicated chromosomes assemble. However, after initiation of anaphase with chromosomal separation and cleavage furrow formation, the furrow regresses, the spindle dissociates, and the megakaryocyte re-enters the G1 phase as a polyploid cell.65,67 Attempts at biochemical analysis of this process have come from leukemic cell lines and normal cultured megakaryocytes. Hypotheses for the mechanisms triggering polyploidy in megakaryocytes have included deficiencies of cyclin B, altered cytoskeletal dynamics, a defect of the chromosomal passenger proteins, and malfunction of the contractile ring. Early interest focused on the M-phase cyclin-dependent kinase cyclin B-cdc2. Also termed mitosis-promoting factor, the amount of cyclin B and the activity of the cyclin B-cdk1 complex rise through the cell cycle until mitosis, where they fall abruptly due to proteosomal degradation of cyclin B. As the major departure from normal cell-cycle behavior in megakaryocytes occurs during the M phase, it was proposed that endomitosis might result from impaired activation of cyclin B-cdc2 kinase, as suggested in several cell line models.68,69 and 70 However, results from normal murine and human megakaryocytes argue for normal mitotic kinase activity during endomitosis. Using TPO to expand primary megakaryocytes in vitro, two groups have demonstrated the presence of functional cyclin B and cdc2 in endomitotic megakaryocytes65,71 as well as a normal metaphase checkpoint mechanism.72 Although the combined deletion of cyclin D1, D2, and D3 in mouse models did not specifically affect megakaryocytes,73 deletion of cyclin E did impair megakaryopoieis as well as trophoblast development,74 suggesting that cyclin E plays a role in endomitosis.
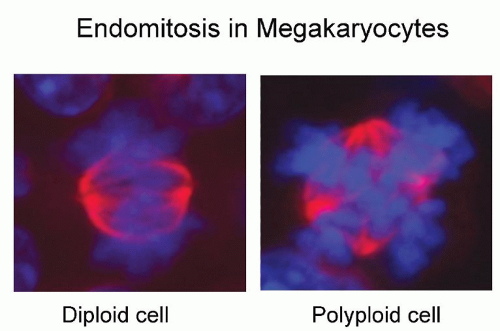 FIGURE 15.3. Endomitosis in megakaryocytes. (A) Mitosis in a megakaryotic precursor: chromatids (blue) are arranged in a single metaphase plate, microtubules (red) form a bipolar spindle. (B) Endomitosis: chromatids are arranged in two metaphase plates and the spindle is multipolar. |
Alterations in regulation of the microtubule cytoskeleton have also been proposed as fundamental in triggering endomitosis. Stathmin is a microtubule-depolymerizing protein that is important for the regulation of the mitotic spindle. As cells enter mitosis, the microtubule-depolymerizing activity of stathmin decreases, allowing microtubules to polymerize and assemble into a mitotic spindle. Reactivation of stathmin in the later stages of mitosis is necessary for the disassembly of the mitotic spindle and the exit from mitosis. Interfering with stathmin expression disrupts the normal mitotic spindle and leads to aberrant mitotic exit.75 Thus it has been hypothesized that alterations in stathmin expression underlie endomitosis in megakaryocytes. In support of this view, expression of stathmin is decreased in higher ploidy megakaryocytes,76 and studies in the erythroleukemia cell line K562 show that inhibition of stathmin expression enhances polyploidy, whereas overexpression of stathmin inhibits the transition from a mitotic cycle to an endomitotic cycle and reduces formation of multipolar mitotic spindles.76,77 Whether down-regulation of stathmin leads to polyploidization in megakaryocytes or is a result of it is not known. Proper activation and localization of
the chromosomal passenger proteins have also been examined as a potential point of departure from the mitotic cell cycle in megakaryocytes. These proteins, including INCENP, survivin, and Aurora Kinase B, form a complex that localizes to the centrosome prior to anaphase and then shifts to the microtubules of the midbody and cleavage furrow.78 Elimination of individual members of the passenger protein complex in organisms as diverse as Drosophila and mammals leads to the abrogation of proper cell division and the formation of a cellular syncytium containing a large nuclear mass.79,80 and 81 Work in cell lines initially suggested that megakaryocytes might be deficient in survivin or Aurora kinase B.82,83 However, subsequent examination of primary megakaryocytes revealed that Aurora kinase B as well as associated passenger proteins are active and appropriately localized in both diploid and polyploid megakaryocytes.84 Further observation of endomitosis by live cell microscopy demonstrated that endomitotic megakaryocytes assemble cleavage furrows that begin to contract and then regress; this fact suggests that the “defect” in endomitosis is beyond anaphase and involves the function of the contractile ring in cytokinesis.85,86 Confocal microscopy confirmed poor localization of RhoA and the actin/myosin complex to the cleavage furrow in endomitotic megakaryocytes. In support of the role of RhoA in endomitosis, expression of Ect2 and GEF-H1, guanine exchange factors that activate RhoA during cytokinesis, is decreased in polyploidizing cells, and forced overexpression of Ect2 and GEF-H1 blocks endomitosis.87 Recent work indicates that nonmuscle myosin IIB heavy chain, whose expression is controlled by Runx1, is also down-regulated during megakaryocyte maturation, and that loss of myosin IIB expression inhibits the return of 4 N cells to the diploid state.88 It is likely that there are multiple alterations in the cell cycle of megakaryocytes that are involved in the process of endomitosis.
the chromosomal passenger proteins have also been examined as a potential point of departure from the mitotic cell cycle in megakaryocytes. These proteins, including INCENP, survivin, and Aurora Kinase B, form a complex that localizes to the centrosome prior to anaphase and then shifts to the microtubules of the midbody and cleavage furrow.78 Elimination of individual members of the passenger protein complex in organisms as diverse as Drosophila and mammals leads to the abrogation of proper cell division and the formation of a cellular syncytium containing a large nuclear mass.79,80 and 81 Work in cell lines initially suggested that megakaryocytes might be deficient in survivin or Aurora kinase B.82,83 However, subsequent examination of primary megakaryocytes revealed that Aurora kinase B as well as associated passenger proteins are active and appropriately localized in both diploid and polyploid megakaryocytes.84 Further observation of endomitosis by live cell microscopy demonstrated that endomitotic megakaryocytes assemble cleavage furrows that begin to contract and then regress; this fact suggests that the “defect” in endomitosis is beyond anaphase and involves the function of the contractile ring in cytokinesis.85,86 Confocal microscopy confirmed poor localization of RhoA and the actin/myosin complex to the cleavage furrow in endomitotic megakaryocytes. In support of the role of RhoA in endomitosis, expression of Ect2 and GEF-H1, guanine exchange factors that activate RhoA during cytokinesis, is decreased in polyploidizing cells, and forced overexpression of Ect2 and GEF-H1 blocks endomitosis.87 Recent work indicates that nonmuscle myosin IIB heavy chain, whose expression is controlled by Runx1, is also down-regulated during megakaryocyte maturation, and that loss of myosin IIB expression inhibits the return of 4 N cells to the diploid state.88 It is likely that there are multiple alterations in the cell cycle of megakaryocytes that are involved in the process of endomitosis.
Another important aspect of megakaryocyte maturation is the development of the DMS. Initially described more than 30 years ago,89 what begins as invaginations of the plasma membrane ultimately becomes a highly branched interconnected system of channels that course through the cytoplasm. The use of electrondense tracers indicated that the DMS is in open communication with the extracellular space,52,89,90 in comparison with the rough endoplasmic reticulum which is contiguous with the nuclear membrane. Over the 72 hours it takes for stage III and stage IV cells to develop from stage I megakaryocytes, the DMS grows by approximately 25-fold. The mature DMS contains phosphatidyl inositol-4,5-P2 (PI-4,5-P2), a phospholipid that is usually associated with the plasma membrane, and PI-4,5-P2 promotes actin polymerization by activating Rho-like guanosine triphosphatases (GTPases) and Wiskott-Aldrich syndrome (WASp) family proteins.91 The purpose of the DMS has been disputed for nearly 20 years92; initially, it was thought to compartmentalize the cytoplasm of the mature megakaryocyte into platelet territories, which ultimately fragment into mature platelets along the cleavage planes so formed, but recent evidence suggests that the DMS provides the necessary membrane required for the formation of proplatelet processes and may promote actin polymerization.91
Two specific types of granules are characteristic of mature megakaryocytes and platelets. The more abundant α-granules first begin to form adjacent to the Golgi apparatus as 300- to 500-nm round or oval organelles in stage I and II cells. α-Granules contain multiple proteins with functions in coagulation and angiogenesis. Three distinct compartments are recognized in α-granules: (a) a central electron-dense nucleoid containing fibrinogen, platelet factor 4 (PF4), β-thromboglobulin, transforming growth factor (TGF)-β1, vitronectin, multimerin, and tissue-type plasminogen activator; (b) a relatively lucent peripheral zone containing tubules and vWF (an arrangement akin to Weibel-Palade bodies found in endothelial cells); and (c) the granule membrane containing many of the critical platelet receptors for cell rolling (P-selectin, also termed CD62p), firm adhesion (gpIb/V/IX), and aggregation (αIIbβ3 integrin). In platelets, proteins present in α-granules arise from de novo megakaryocyte synthesis (e.g., gpIb/V/IX, gpIV, αIIbβ3 integrin, vWF, P-selectin, β-thromboglobulin, and plateletderived growth factor), nonspecific pinocytosis of environmental proteins (e.g., albumin and IgG), or cell surface membrane receptor-mediated uptake from the environment (e.g., fibrinogen, fibronectin, and factor V).93,94 Dense granules are less numerous and contain the vasoconstrictive 5-hydroxytryptamine, the nonmetabolic pool of adenosine triphosphate and adenosine diphosphate (ADP), calcium, and magnesium, and they have a fundamental role in hemostasis. Although α-granules contain multiple proteins with opposing functions, proteins with specific functions may be released selectively in response to certain agonists. For example, ADP and PAR1 stimulate the release of angiogenic factors such as VEGF, whereas thromboxane A2 and PAR4 stimulate the release of antiangiogenic factors such as endostatin.95,96 Therefore, current models propose differential secretion of granule contents in response to different agonists, although the mechanisms by which this selectivity is achieved are still unclear.
Throughout megakaryocyte development, the cytoplasm acquires a rich network of microfilaments and microtubules. Toward stages III and IV, these proteins accumulate in the cell periphery, creating an organelle-poor peripheral zone. Biochemically, the megakaryocyte cytoskeleton is composed of actin, α-actinin, filamin, nonmuscle myosin, β1 tubulin, talin, and spectrin. Spectrin plays an important role in stabilizing the DMS and proplatelet processes.97 β1 tubulin is expressed exclusively in megakaryocytes and platelets and is critical for proplatelet formation; over 90% of microtubules in the mature megakaryocyte are composed of the β1 isoform.98,99 At the onset of proplatelet formation, blunt pseudopodia are formed, and β1-tubulin-containing microtubules assemble into thick bundles below the plasma membrane100 (Fig. 15.4). The pseudopodia elongate, bend, and branch to form proplatelets, and the microtubules form bidirectional linear arrays with terminal loops composed of 8 to 12 microtubule coils at the proplatelet tips.101 Dynein powers microtubule sliding and proplatelet elongation, whereas actin and myosin are involved in proplatelet bending and branching.100,102 Granules and organelles such as mitochondria traffic on these microtubule “tracks” to accumulate within the tips that will ultimately be released to become circulating platelets.103 The coiled microtubules at the proplatelet tip become the marginal band that maintains the discoid shape of the platelet.100,104 RanBP10 binds to microtubules and functions as a GEF for Ran, stabilizing microtubules and facilitating granule transport.105,106 Remodeling of the megakaryocyte cytoskeleton is essential for proplatelet formation, and mutations affecting cytoskeletal proteins, including regulators of actin (WASp), filamin A, β1-tubulin, and nonmuscle myosin heavy chain IIA form the basis of several of the inherited thrombocytopenia syndromes.55,107,108,109
Platelets are derived from the megakaryocyte cytoplasm. It has been estimated that each megakaryocyte gives rise to 1,000 to 5,000 platelets110,111 and 112 before the residual nuclear material is engulfed and eliminated by marrow macrophages.113 Stage IV megakaryocytes are wholly engaged in platelet formation. Careful microscopic studies have localized marrow megakaryocytes to the abluminal surface of sinusoidal endothelial cells. Fully mature megakaryocytes develop cytoplasmic processes constricted at platelet-sized intervals that extend through the endothelial barrier into the sinusoidal lumen, where platelets are released.114 It is likely that both integrin-mediated cell-cell interactions and extracellular matrix degradation are required for this process.115,116 and 117 It is also possible that the final stages of megakaryocyte fragmentation occur in the lung, at least for some megakaryocytes. Howell and Donahue reported in 1939 that platelet levels in pulmonary venous blood exceed those found in the pulmonary artery, suggesting platelet production in the lung.118 Whether this represents the migration and fragmentation of intact megakaryocytes in
the lung or merely the final size reduction of large fragments of megakaryocyte cytoplasm that are also released into the blood119 is not clear. Lung megakaryocytes have been characterized by Slater et al., who believe they contribute substantially to blood platelet production.120 However, in preclinical studies of TPO, Kaushanksy et al. did not detect any denuded megakaryocyte nuclei in the lungs of mice which at the time of sacrifice had platelet counts as high as 4 million/mm3.121 One study found that dog lungs contain 2.5 megakaryocytes/cm2 tavassoli122; extrapolation of these data would suggest that human lungs contain approximately 6,000 megakaryocytes, only enough to account for a small proportion (<0.1%) of daily platelet production. Moreover, cultured megakaryocytes can form functional platelets in vitro by generating proplatelets in the absence of endothelial surfaces or the pulmonary circulation,100 and the release of proplatelet fragments into bone marrow sinusoids has been visualized in living mice.123 Thus, the role of pulmonary bed platelet production remains controversial.
the lung or merely the final size reduction of large fragments of megakaryocyte cytoplasm that are also released into the blood119 is not clear. Lung megakaryocytes have been characterized by Slater et al., who believe they contribute substantially to blood platelet production.120 However, in preclinical studies of TPO, Kaushanksy et al. did not detect any denuded megakaryocyte nuclei in the lungs of mice which at the time of sacrifice had platelet counts as high as 4 million/mm3.121 One study found that dog lungs contain 2.5 megakaryocytes/cm2 tavassoli122; extrapolation of these data would suggest that human lungs contain approximately 6,000 megakaryocytes, only enough to account for a small proportion (<0.1%) of daily platelet production. Moreover, cultured megakaryocytes can form functional platelets in vitro by generating proplatelets in the absence of endothelial surfaces or the pulmonary circulation,100 and the release of proplatelet fragments into bone marrow sinusoids has been visualized in living mice.123 Thus, the role of pulmonary bed platelet production remains controversial.
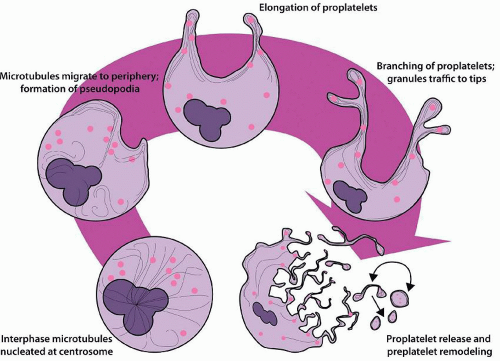 FIGURE 15.4. Proplatelet formation in mature megakaryocytes. Noncentrosomal microtubules migrate to the cell periphery where they form bundles. Pseudopodia form and elongate into proplatelet processes, and the microtubules form bidirectional tracks within the processes that terminate in coiled ends. Granules and other organelles traffic along the microtubules and accumulate in the tips. Proplatelet processes are released and continue to remodel in the circulation, alternating between barbell-shaped proplatelets and large preplatelets, before the proplatelets finally divide into individual platelets. |
Notably, the proplatelets and platelets released in experimental models are larger than typical circulating platelets. In addition, it has long been recognized that patients with disorders of increased platelet production, such as immune thrombocytopenia, have relatively large circulating platelets. Thus, it has been proposed that once released into the circulation, these larger platelet precursors undergo further remodeling.124,125,126 Using an in vitro model, Thon et al. demonstrated that platelet precursors shift back and forth between two morphologic states, discoid preplatelets and barbell-like proplatelets.126 Spectrin stabilizes the bridge between the ends of the barbell form.97 Furthermore, these precursor platelets can be observed to divide or undergo fission. Shear forces in the circulation likely contribute to the cleavage of barbell-shaped proplatelets into individual platelets, although it has been postulated that mechanisms similar to those driving cell separation in cytokinesis may also be involved.
Programmed cell death represents not only the end of the megakaryocyte life cycle and proteins that regulate the intrinsic apoptosis pathway influence megakaryocyte survival. Apoptosis has also been proposed to play a role in megakaryocyte functions such as proplatelet formation. Early studies found that focal activation of caspases accompanies megakaryocyte maturation and overexpression of Bcl2 inhibits proplatelet formation, suggesting that localized apoptosis is required for platelet shedding.127 However, a recent study found that targeted deletion of caspase 9, the initiator caspase in the intrinsic apoptotic pathway, has no effect on proplatelet formation.128 Additional data support the alternative hypothesis that inhibition of apoptosis in mature megakaryocytes is necessary for proplatelet formation. In mouse models, targeted loss of Bcl-xL in megakaryocytes leads to caspase activation and failure of proplatelet formation and release of morphologically abnormal, large platelets.129 Whereas isolated loss of Bcl-xL primarily affects mature megakaryocytes at the point of proplatelet formation, combined deletion of Bcl-xL
and Mcl-1 has even more profound effects, with severe impairment of megakaryopoiesis in embryos lacking both apoptotic regulators.130 In addition, Bcl-xL inhibits apoptosis in circulating platelets. Mutation or pharmacologic inhibition of Bcl-xL leads to reduced platelet life span in mice.129,131 Apoptosis of aged platelets is mediated by Bak and Bax, whose activity is normally controlled by Bcl-xL; genetic deletion of Bak and Bax prolongs platelet survival and renders platelets resistant to the effects of Bcl-xL inhibition.129 Mutations of Bcl-xL or Mcl-1 have not been described in humans; however, a proapoptotic mutation of cytochrome c has been identified in a family with autosomal dominant thrombocytopenia.132
and Mcl-1 has even more profound effects, with severe impairment of megakaryopoiesis in embryos lacking both apoptotic regulators.130 In addition, Bcl-xL inhibits apoptosis in circulating platelets. Mutation or pharmacologic inhibition of Bcl-xL leads to reduced platelet life span in mice.129,131 Apoptosis of aged platelets is mediated by Bak and Bax, whose activity is normally controlled by Bcl-xL; genetic deletion of Bak and Bax prolongs platelet survival and renders platelets resistant to the effects of Bcl-xL inhibition.129 Mutations of Bcl-xL or Mcl-1 have not been described in humans; however, a proapoptotic mutation of cytochrome c has been identified in a family with autosomal dominant thrombocytopenia.132
TRANSCRIPTIONAL REGULATION OF MEGAKARYOPOIESIS
Several themes are emerging in models of transcriptional regulation of hematopoietic differentiation that are relevant to the discussion of megakaryopoiesis. Lineage priming refers to the concept that the HSC is competent to express multiple genetic programs and lineage choice is the consequence of progressive silencing of alternative possibilities.133,134 Similarly, anticipatory regulation refers to the concept that differentiation-associated transcription factors are already bound at target promoters in HSCs, allowing for low-level expression that increases with differentiation.23 Thus, as previously discussed, several transcription factors associated with megakaryocytes, as well as other lineages, are also expressed at low levels in HSCs, as well as in bipotential megakaryocytic/erythroid cells. As cells start to differentiate, lineage-specific transcription factors promote their own expression while suppressing factors that would favor other cell types, thus reinforcing lineage choice. For example, GATA1 activates its own promoter to increase its expression, and simultaneously inhibits PU.1, favoring megakaryocytic/erythroid differentiation, whereas in other cells, PU.1 might dominate, repressing the megakaryocytic/erythroid program and favoring myelopoiesis.135,136,137 Transcription factors do not act in isolation, however, but are typically present as components of cross-regulatory multiprotein complexes; their function may vary depending on the other factors that are present.
Recent studies have identified five core transcription factors, SCL/TAL1, GATA1, GATA2, RUNX1, and FLI1, as the promoters of hematopoietic genes in primary megakaryocytes. Genes next to regions bound by all five factors were highly enriched for known regulators of megakaryocytic differentiation or function.138 Additional factors of importance in megakaryopoiesis include NF-E2, FOG1, HOX family proteins, GFI-1b, and c-Myb. Mutant forms of several of these factors or their downstream targets have been found in inherited thrombocytopenia syndromes or leukemias. In addition, there is emerging recognition of the role of noncoding RNAs in megakaryocytic differentiation.
SCL/TAL1: Stem cell leukemia/T-cell acute lymphoblastic leukemia gene 1, or SCL/TAL1, is a basic helix-loop-helix transcription factor that is essential for early hematopoietic specification as well as definitive differentiation of the erythroid and megakaryocyte lineages.139 Loss of Scl/Tal1 expression in adult mice leads to defects in megakaryocyte cytoplasmic maturation and thrombocytopenia.140,141 In addition, progenitor cells from Scl/Tal1-deficient mice are unable to generate megakaryocytes in vitro.142,143 Among the transcriptional targets of SCL/TAL1 important for megakaryopoiesis are NF-E2 and MEF2C. Scl/Tal1 directly binds regulatory elements of Mef2C in megakaryocytic cells, and the platelet phenotype of Mef2C– knockout mice is similar to that of Scl/Tal1-deficient animals.144 In human CD34+ hematopoietic progenitor cells, expression of MEF2C increases with megakaryocytic differentiation.145 SCL/TAL1 has multiple partners, including E2A and LMO2. E2A binds canonical DNA elements called E-boxes represented by the motif CANNTG. E-boxes are frequently found along with GATA-binding motifs within the regulatory elements of erythroid and megakaryocytic cells.146,147 LMO2 is a LIM domain containing protein that forms a multiprotein complex with GATA1, SCL/TAL1, E2A, and another LIM domain protein LDB1146,148,149 and 150; the complex is critical for early hematopoietic specification.139,148,151 LDB1 can self-associate to form oligomers and may bind multiple GATA1 molecules simultaneously, thus facilitating long-range interactions between GATA-bound sites.152,153 and 154
GATA1: GATA1 is a zinc finger transcription factor critical in both erythroid and megakaryocytic development, with target genes including HBB (encoding β-globin), ALAS1, BCL2L1, NFE2, GP1BB, and GP9. Gata1 deficiency in mouse models leads to arrest of erythroid maturation at the proerythroblast stage and severe anemia,155 as well as defects in megakaryocyte maturation with hyperproliferation of low ploidy cells, impaired endomitosis and formation of the DMS, and release of large hypogranulated platelets.156,157 GATA-binding motifs (T/A)GATA(A/G) are found in the regulatory elements of nearly all erythroid and megakaryocytic genes.158,159 Furthermore, the co-occurrence of ETS and GATA motifs was significantly associated with expression of the megakaryocytic program over the erythroid program.160 GATA1 can function either as a transcriptional activator or repressor, depending on its binding partners. GATA1 contains two zinc fingers; the C-terminal zinc finger is responsible for high-affinity DNA binding, whereas the N-terminal finger stabilizes the interaction and binds to the co-factor FOG1.161 In addition to FOG1,162 GATA1 has interactions with other megakaryocytic core factors, including RUNX1 and FLI1.163,164,165 FOG1 contains 9 zinc fingers, 4 of which mediate interactions with GATA1.166 In mouse models, deletion of Fog1 results in a more severe defect in megakaryopoiesis than does deletion of Gata1,167,168 supporting the conclusion that although GATA1 is required for erythroid lineage commitment, FOG1 is required for specification of both erythroid and megakaryocytic progenitors.168 The interaction of GATA1 with FOG1 and PU.1 is mutually exclusive,169 thus GATA1 bound by FOG1 may be protected from repression by PU.1, favoring megakaryocytic/erythroid differentiation. Posttranslational modification of GATA1 by SUMOylation of amino acid K137 also disrupts FOG1 binding.170 FOG1 can promote the function of GATA1 either as an activator or repressor of transcription. FOG1 contains an N-terminal nucleosome remodeling and histone deacetylase complex (NuRD) recruitment domain,171 and can thus bind transcriptional repressors such as HDAC1 and HDAC2. Knockin mice expressing FOG1 mutated within its NuRD domain exhibited thrombocytopenia and anemia, suggesting that FOG1 function in megakaryopoiesis is mediated at least in part through recruitment of NuRD.172 Although association with NuRD was linked with both gene activation and repression, NuRD binding by FOG1 was shown to repress the mast cell program in megakaryocytic/erythroid cells.173 In humans, X-linked inheritance of mutations of GATA1 involving the N-zinc finger, abrogating interaction with FOG1, leads to macrothrombocytopenia and dyserythropoiesis174,175,176,177 and 178 (Fig. 15.5). In Down syndrome, acquired mutations within exon 2 of GATA1 result in alteration of the translational start site and production of a shortened species, GATA1S. GATA1S binds DNA and interacts with FOG1, but lacks the N-terminal activation domain and has a reduced transactivation potential.179 GATA1S is nearly always present in children with Down syndrome who develop transient abnormal myelopoiesis and megakaryoblastic leukemia and is thought to be an early event in the initiation of leukemogenesis.180,181
GATA2: Closely related to GATA1, GATA2 is an essential transcription factor for HSCs, MEPs, and megakaryocytes, but becomes down-regulated in mature erythroid cells.182,183,184 and 185 GATA2 expression is down-regulated to a lesser extent during
megakaryopoiesis,186,187 and overexpression of GATA2 favors megakaryopoiesis in cell lines.188 In maturing cells, expression of GATA2 is repressed by GATA1, reinforcing the differentiation program.189 Thus, in experimental models, deletion of GATA1 is accompanied by overexpression of GATA2.190 GATA1 and GATA2 have homologous zinc fingers, interact with FOG1, and bind overlapping sets of genes.191,192 However, in general, GATA1 and GATA2 have opposite effects at target genes, either activating or repressing transcription. The exchange of GATA2 for GATA1 at regulatory elements during differentiation is referred to as the GATA switch.193 Using an in vitro model of megakaryocyte maturation, Dore et al. recently found that as many as 30% of sites occupied by GATA2 are occupied instead by GATA1 in differentiated cells.160 In contrast to GATA1 which is on the X-chromosome, GATA2 is on chromosome 3 and GATA2 mutations have not been described in human disease.
megakaryopoiesis,186,187 and overexpression of GATA2 favors megakaryopoiesis in cell lines.188 In maturing cells, expression of GATA2 is repressed by GATA1, reinforcing the differentiation program.189 Thus, in experimental models, deletion of GATA1 is accompanied by overexpression of GATA2.190 GATA1 and GATA2 have homologous zinc fingers, interact with FOG1, and bind overlapping sets of genes.191,192 However, in general, GATA1 and GATA2 have opposite effects at target genes, either activating or repressing transcription. The exchange of GATA2 for GATA1 at regulatory elements during differentiation is referred to as the GATA switch.193 Using an in vitro model of megakaryocyte maturation, Dore et al. recently found that as many as 30% of sites occupied by GATA2 are occupied instead by GATA1 in differentiated cells.160 In contrast to GATA1 which is on the X-chromosome, GATA2 is on chromosome 3 and GATA2 mutations have not been described in human disease.
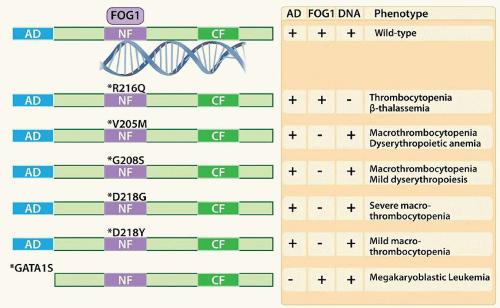 FIGURE 15.5. GATA1 mutations associated with human disease. Wild-type GATA1 contains an activation domain (AD) and two zinc fingers (NF and CF) that mediate DNA binding. FOG1 interacts with the amino-terminal zinc finger (NF). Mutations in the N-finger have been discovered in males with macrothrombocytopenia and impaired erythropoiesis, either due to disruption of FOG1 binding or, in the case of R216Q, impaired DNA binding. In Down syndrome, mutations that lead to loss of the AD (GATA1S) promote the development of transient myeloproliferative disorder and megakaryoblastic leukemia. |
RUNX1: RUNX1 is a heterodimeric transcription factor of the Runt-domain family, consisting of core-binding factor α-2 (CBFA2) that binds DNA and CBF-β, which does not directly bind DNA.194 Although RUNX1 is critical for emergence of the HSC during embryogenesis, in the adult it is dispensable for maintenance of stem cells but required for megakaryopoiesis.195 In cell line and animal models, overexpression of RUNX1 induces megakaryocytic differentiation, whereas RUNX1 deficiency results in proliferation of immature megakaryocytes and thrombocytopenia.163,195,196 Mutations leading to haploinsufficiency of RUNX1 cause familial thrombocytopenia with defective megakaryocyte maturation and a striking predisposition to myeloid leukemias.197 Targets of RUNX1 in megakaryopoiesis include c-Mpl.198 RUNX family members frequently cooperate with co-factors such as CCAAT box/enhancer-binding protein (C/EBP)α, ETS family members, p300/CBP, and c-Myb.199,200 In vitro models have demonstrated cooperation of RUNX1 with GATA1 and ETS family members in the activation of megakaryocytic target genes.163,165,201,202 RUNX1 and GATA1 also modulate the activity of the cyclin-dependent kinase positive transcription elongation factor (P-TEFb), thus affecting transcriptional elongation203; induction of megakaryocytic differentiation correlates with P-TEFb activity.204
NF-E2: NF-E2 is a heterodimeric transcription factor containing a p45 leucine-zipper subunit and a p18 subunit MafG. NF-E2 binds DNA through Maf-recognition elements, but p45 is required for transcriptional activation.205 NF-E2 is essential for the final differentiation of megakaryocytes and for platelet formation, as p45−/− mice are severely thrombocytopenic (≤5% the normal platelet count) despite increased numbers of megakaryocytes.206,207 Several target genes of NF-E2 have been identified, including TUBB1 (encoding β1-tubulin), CASP12 (caspase 12), TBXAS1 (thromboxane A synthase-1), RAB27B, and 3β-HSD 3β-hydroxysteroid dehydrogenase, a mediator of autocrine biosynthesis of estradiol within megakaryocytes.99,208,209,210 and 211 Intriguingly, ectopic expression of 3β-HSD in NF-E2-deficient megakaryocytes rescues proplatelet formation.212 Platelets in NF-E2−/− mice are spherocytic, due to loss of the integrity of the marginal band, normally comprised of β1-tubulin.208 NF-E2 is itself a target of GATA1 and SCL/TAL1.141
Homeobox Genes: The homeobox genes, which include HOX and non-HOX genes, play key roles in body pattern development, and they are also expressed in adult HSC and play pivotal roles in their proliferation. In hematopoietic progenitors, TPO promotes expression of HOXB4 as well as nuclear localization of HOXA9 through up-regulation of its co-factor MEIS1.37,38 In addition to HOXA9, MEIS1 can interact with PBX1b and PBX2, and the
complex of MEIS1 and PBX up-regulates megakaryocytic gene expression in cooperation with GATA1 and ETS.213 Meis1−/− mice have a severe abnormality of megakaryopoiesis and lack CD41+ cells in the fetal liver.214,215 Although its function in megakaryocytes is not understood, mutations of HOXA11 are associated with the congenital thrombocytopenia syndrome amegakaryocytic thrombocytopenia with radioulnar synostosis (ATRUS).216
complex of MEIS1 and PBX up-regulates megakaryocytic gene expression in cooperation with GATA1 and ETS.213 Meis1−/− mice have a severe abnormality of megakaryopoiesis and lack CD41+ cells in the fetal liver.214,215 Although its function in megakaryocytes is not understood, mutations of HOXA11 are associated with the congenital thrombocytopenia syndrome amegakaryocytic thrombocytopenia with radioulnar synostosis (ATRUS).216
FLI1, KLF1, and c-Myb: Several changes in gene expression accompany the divergence of bipotential MEPs to committed megakaryocytic or erythroid cells. The ETS family member FLI1, in conjunction with SCL and GATA2, controls HSC specification217; however, it is also critical for formation of the vasculature and for megakaryopoiesis.218,219,220 and 221 FLI1 cooperates with GATA1222,223 and regulates many genes important for terminal megakaryocytic maturation.222,224 Expression of FLI1 influences cell fate decisions in the MEP; increased expression of FLI1 favors megakaryocyte development, whereas FLI1 expression decreases in erythroid cells. In mouse models, loss of Fli1 suppresses megakaryopoiesis, whereas overexpression of Fli1 promotes megakaryocyte development while suppressing erythropoiesis.218,225,226 Conversely, KLF1 (or EKLF), a target of GATA1, is up-regulated in erythroid-committed progenitors, but inhibits megakaryopoiesis.227,228,229 FLI1 and KLF1 inhibit each other’s activity directly through protein-protein interaction,229 and this cross-antagonism has been suggested to reinforce lineage choice between megakaryocytes and erythroid cells.168 Indeed, selective disruption of Klf1 in mouse models leads to anemia with thrombocytosis,230 whereas targeted deletion of Fli1 enhances erythropoiesis.231 Other transcription factors have been identified that act in the MEP, including GFI-1b and c-Myb. Growth factor independence-1B (GFI-1b) is a transcriptional repressor that is essential for both erythropoiesis and megakaryopoiesis.232,233 GFI-1b is expressed in the bipotent MEP and inhibits the expression of the TGF-β receptor III (TGFBR3) gene. This disrupts TGF-β/smad2 signaling which otherwise restrains proliferation and differentiation of erythro-megakaryocytic progenitors.234 The c-Myb gene encodes for a basic helix turn helix transcription factor composed of three functional domains: a DNA binding domain at the N terminus, a central transactivation domain, and a C-terminal negative regulatory domain. c-Myb has critical roles both during early hematopoiesis and in differentiating cells.235,236 Down-regulation of c-Myb accompanies megakaryocytic differentiation, and hypomorphic c-Myb expression increases megakaryopoiesis at the expense of erythropoiesis.236,237 Silencing of c-Myb in human CD34+ progenitor cells in vitro is associated with loss of KLF1 and LMO2 (http://bloodjournal.hematologylibrary.org/content/116/22/e99.full, ref 22) expression and results in commitment toward the monomacrophage and megakaryocyte lineages whereas erythroid differentiation is strongly impaired.238
Recently, the role of noncoding RNAs has been recognized in hematopoiesis and megakaryocyte differentiation.239,240,241 MicroRNAs (miRNAs) are a class of approximately 22-nucleotide noncoding RNAs that have been highly conserved during evolution and modulate protein expression by degrading mRNA or repressing translation. There are over 400 human miRNAs, and several of them have been implicated in megakaryopoiesis using profiling, knockdown, or overexpression assays. In megakaryocytes, miR-155 expression is reduced as cells differentiate toward megakaryocytes, and overexpression of miR-155 inhibits megakaryopoiesis in in vitro and mouse models, potentially by targeting Meis1 and Ets1 transcripts.242,243 and 244 In contrast, miR-150 levels increase during megakaryocyte differentiation, but not in erythroid cells.239 miR-150 targets c-Myb expression. Down-regulation of c-Myb promotes megakaryocytic differentiation at the expense of erythroid cells.236 TPO induces expression of miR-150, providing another mechanism by which it influences megakaryopoiesis.245 Additional regulatory miRNAs with potential roles in megakaryopoiesis include miR-34a which when overexpressed in CD34+ cells increases megakaryocyte colony formation, and miR-27a which targets the Runx1 transcript.246,247
CYTOKINES IN MEGAKARYOPOIESIS
The identification of TPO and its receptor c-Mpl was a major advance in the study of megakaryocytes. The term thrombopoietin was first coined in 1958 to describe the putative primary regulator of platelet production248; the anticipated factor was also referred to as megakaryocyte growth and development factor (MGDF). Over the four decades following its description, evidence both for and against the existence of TPO as a distinct entity was presented.249,250 Although not appreciated at the time, a major insight into megakaryocyte biology was provided in 1986 when Françoise Wendling et al. described a murine retrovirus, myeloproliferative leukemia virus, which induced a vast expansion of hematopoietic cells.251 The responsible viral oncogene was characterized in 1990,11 and its cellular homolog c-mpl was cloned in 1992.10 Based on the presence of two copies of the hematopoietic cytokine receptor motif, it was immediately apparent that c-mpl encoded a growth factor receptor. However, its ligand was not known. Using three distinct strategies, four separate groups were able to clone the complementary DNA for the corresponding hormone and reported their results in 1994.12,13,14 and 15 The gene for this c-Mpl ligand was mapped to human chromosome 3q26-28, spans 6 to 8 kb, consists of seven exons, and encodes a predicted 36-kD polypeptide.252,253 and 254 The recombinant and naturally occurring cytokines migrate as 70-kD proteins, indicating substantial posttranslational modification, a conclusion verified by the finding of six sites of N-linked carbohydrate and many sites of O-linked carbohydrate.255 Based on its biologic activities, the c-Mpl ligand was termed thrombopoietin (TPO).121
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


