FIGURE 42-1. The upper panel of the figure lists the density range that encompasses each lipoprotein class and the major tissue in which the lipoprotein is synthesized. The bottom panel is a graphic representation of a generic lipoprotein, emphasizing the spherical shape of the particle, the use of amphipathic apoproteins and polar lipids on the particle surface that interact with cells and the blood environment, and the sequestration of nonpolar lipids in the core of the lipoprotein. All lipoproteins share these general features, but they differ in size, lipid composition, and in the specific apoproteins embedded.
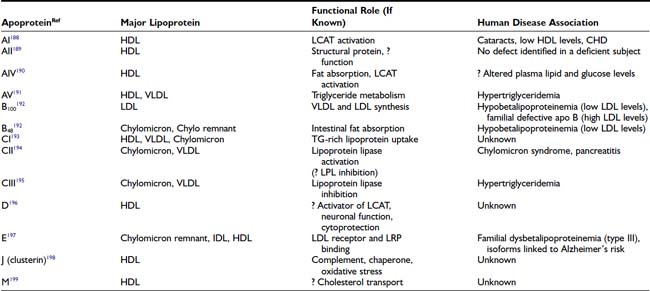
The nomenclature for most lipoprotein particles derives from the method by which they were originally identified and separated, that is, density gradient ultracentrifugation. For a brief period after the introduction of gel electrophoresis, lipoproteins were also classified by their migration properties on these gels, giving rise to alternative names such as alpha and beta lipoproteins. This period coincided with the identification of many of the clinical syndromes caused by abnormalities of lipid metabolism, so the names for some of these disorders still carry the signature of the electrophoresis era (e.g., hypoalphalipoproteinemia indicates low levels of the alpha-migrating or high-density lipoprotein [HDL]). Gel electrophoresis is rarely used in clinical laboratories today for standard lipoprotein measurements, and so the medical literature has largely returned to the use of the density nomenclature to describe and characterize disorders of lipoprotein metabolism.
Although lipoproteins constitute a continuous spectrum of density within the plasma, they are commonly divided into five major classes (see Fig. 42-1). These are: (1) chylomicrons; (2) very low-density lipoproteins (VLDL); (3) intermediate-density lipoproteins (IDL); (4) low-density lipoproteins (LDL); and (5) high-density lipoproteins (HDL). Within these classes, further subdivision of lipoproteins can be made. In diabetics, a higher proportion of LDL is carried in a denser, smaller-diameter subfraction that is called small dense LDL (sdLDL). HDL are commonly divided into HDL2 and HDL3 subclasses, but some investigators have adopted a gel separation method to segregate HDL into a much larger number of subtypes.6 It is important for the clinician to understand that the different laboratory methods of identifying subpopulations of lipoproteins yield different or overlapping species of particles, causing confusion for those not conversant with the separation techniques. In this chapter, the most widely used classifications will be employed for simplicity, but further research may prove that more sophisticated subclassifications have considerable clinical relevance. Overall, it is helpful to remember that the least dense of the lipoproteins are also the largest in size and the most triglyceride-rich. These are the chylomicrons and VLDL. IDL contain substantial amounts of both triglyceride and cholesterol, whereas the LDL and HDL are predominantly cholesterol-carrying particles with very little associated triglyceride. This information permits the clinician to identify the most likely lipoprotein abnormalities when presented with a patient’s total serum triglyceride and cholesterol values. A brief description of each of the five major lipoprotein classes is summarized in the following paragraphs. A reader interested in a more comprehensive introduction to lipoprotein metabolism should consult the lipid sections of the Metabolic Basis of Inherited Disease.7
Chylomicrons
Chylomicrons derive from dietary fat and carry triglycerides throughout the body. The major structural protein of chylomicrons is apolipoprotein (apo) B48, a protein that is produced from apo B100 RNA by a unique editing process employed by gut cells that generates a protein that is 48% of the length of apo B100.8 Chylomicrons also contain other apoproteins, including members of the apo C and apo A family. These proteins can redistribute to other lipoprotein classes within serum as chylomicrons transfer their lipid content to cells and interacting lipoproteins. Chylomicrons have the lowest density of all lipoproteins and will float to the top of a plasma specimen left in a refrigerator overnight, forming a creamlike layer. Owing to its large size, the chylomicron does not readily enter the artery wall and is therefore thought not to be atherogenic, but the atherosclerotic role of the triglyceride-depleted chylomicron remnant remains controversial. Triglyceride makes up most of the chylomicron and is removed by the action of an enzyme that is bound to the surface of endothelial cells, lipoprotein lipase (LPL). Patients deficient in this enzyme or its apoprotein cofactor (apo CII) have very high serum triglyceride levels and are at increased risk for developing acute pancreatitis. Inasmuch as insulin is also a critical cofactor for LPL activity, the vast majority of patients presenting with hypertriglyceridemic pancreatitis have poorly controlled diabetes as the cause of their delayed chylomicron clearance.
Very Low-Density Lipoproteins
VLDLs are also triglyceride-rich, but their triglyceride content is lower and cholesterol content higher than that of chylomicrons. The protein composition of VLDL also differs from chylomicrons in that the major structural protein is full-length apo B (apo B100) as opposed to the truncated apo B48 form. Like chylomicrons, VLDLs are substrates for lipoprotein lipase–mediated triglyceride removal. Their function is to carry triglycerides synthesized in the liver and intestines to capillary beds in adipose tissue and muscle, where they are hydrolyzed to provide fatty acids that can be oxidized to produce adenosine triphosphate (ATP) for energy production. Alternatively, if not needed for energy production, they can be reesterified to glycerol and stored as fat. After removal of their triglyceride, VLDL remnants (called IDLs) can be further metabolized to LDL. VLDLs serve as acceptors of cholesterol transferred from HDL, accounting in part for the inverse relation between HDL cholesterol and VLDL triglyceride. This transfer process is mediated by an enzyme called cholesterol ester transfer protein (CETP). The most common lipid abnormality seen in type 2 diabetics is an elevated VLDL level, the causes of which will be discussed in the section on diabetic lipid abnormalities.
Low-Density Lipoproteins
LDLs are the major carriers of cholesterol in humans, responsible for supplying cholesterol to tissues with the highest sterol demands. LDLs are also the lipoproteins most clearly implicated in causing atherogenic plaque formation.9 Circulating LDL levels can be increased in persons who consume large amounts of saturated fat and/or cholesterol.10 LDL levels are also elevated in those who have genetic defects that affect LDL receptor function (familial hypercholesterolemia, PCSK9 mutations, autosomal recessive hypercholesterolemia), the structure of LDL’s apoprotein, apo B, or who have polygenic disorders affecting LDL metabolism.11 Circulating LDLs are taken up by the endothelium lining the artery wall, traverse it, and are deposited and trapped in the arterial intima. There, they may undergo oxidation or other biochemical modification, be taken up by macrophages, and stimulate atherogenesis. The association of total serum cholesterol with CHD is predominantly a reflection of the role of LDL. Studies in patients with diabetes have produced somewhat conflicting data on the prevalence of increased LDL levels, as defined by general population norms, but the preponderance of the evidence indicates that diabetics have LDL levels that are similar to those seen in well-matched, nondiabetic control populations. Given diabetics’ greater risk of developing coronary artery disease (CAD), however, the National Cholesterol Education Program guidelines now define LDL levels above 100 mg/dL as being undesirable in diabetics. When judged by this stringent criterion, elevated LDL levels are extremely common in patients with diabetes. In addition, the LDL in diabetics is frequently smaller, denser, and more readily oxidized—properties that are associated with a higher degree of atherogenicity.12,13
High-Density Lipoproteins
HDLs are the smallest of the lipoproteins; despite this, they carry a variety of apoproteins, including members of the apo A and C family. HDLs, unlike the other lipoproteins, are believed to function in humans primarily to return lipids from peripheral tissues to the liver and gut for excretion, rather than moving lipids from the gut organs to the periphery.14 This perspective on HDL function is an oversimplification, since cell culture experiments indicate that HDL can donate lipids as well as pick them up. It has proven to be quite difficult to quantitate the net movement of cholesterol carried in HDL in and out of peripheral tissues in whole animals. Further complicating the issue is the fact that many of the standard laboratory animals used in metabolic studies carry most of their serum cholesterol in HDL rather than LDL, diminishing the relevance of their lipoprotein metabolism to that of the LDL-rich human. Nevertheless, a considerable body of evidence suggests that HDL functions to protect tissues from unwanted accumulation of cholesterol. The mechanisms by which HDL can protect against atherosclerosis include its involvement in the reverse cholesterol transport pathway described earlier, as well as through contributions arising from its carriage of proteins that appear to have antiinflammatory and antioxidant properties.15,16 The unesterified cholesterol from tissues that is transferred to HDL is esterified by the action of lecithin cholesterol acyltransferase (LCAT) and stored in the central core of HDL. This esterified cholesterol can be transferred back to lower-density lipoproteins by the action of cholesterol ester transfer protein, or it may be removed at the liver by the action of a plasma membrane receptor called scavenger receptor B-1 (SRB-1). A particularly effective reverse transport system is thought to explain, at least in part, the association of elevated HDL cholesterol levels with a reduced risk of developing CHD. Apolipoprotein AI (apo AI) is the major apoprotein of HDL, and its serum concentration also correlates inversely with the risk of CHD. Women have higher levels of HDL cholesterol than men, and this may partly explain the lower incidence of CHD in premenopausal women. Exercise increases HDL levels; obesity, hypertriglyceridemia, and smoking lower them. Patients with diabetes, having higher body mass indices and hypertriglyceridemia, typically have lower HDL cholesterol levels than do nondiabetics, contributing further to the atherogenic lipid profile of the diabetic.
METABOLISM OF LIPIDS AND LIPOPROTEINS
The synthesis of lipoproteins in the liver and gut is a complex process regulated by hormonal and transcriptional networks that are affected by dietary intake of both lipid and carbohydrate. The disorders of carbohydrate and lipid metabolism caused by diabetes have both direct and indirect effects on lipoprotein metabolism. A brief summary of normal lipoprotein metabolism will be presented in this section and followed by a description of the impact of diabetes on these processes in the next section.
Following ingestion of a meal containing fat, the triacylglycerols (TAG) are broken down by pancreatic triacylglycerol lipase to yield sn-2-monoacylglycerol and fatty acids. Ingested cholesterol is solubilized by bile salts into micellar structures, a step which is disrupted by bile acid resin binders. The lipids then transverse the enterocyte brush border membrane and are reassembled in the intestinal cell into a triglyceride-rich lipoprotein, the chylomicron. The role of transport proteins in the passage across the enterocyte membrane remains disputed, with evidence for both a passive diffusion and an active transport mechanism.17,18 For cholesterol, a second transport step translocates the sterol into intracellular pools that can be accessed for lipoprotein packaging.19 Niemann-Pick C1-like 1 protein (NPC1L1) appears to be required for this second step to occur and is the target of the cholesterol absorption inhibitor, ezetimibe.20,21 The internalized cholesterol is moved to the endoplasmic reticulum where the monoacylglycerol and fatty acids are also carried by transport proteins. The latter are recombined into triacylglycerol and bound by microsomal triglyceride transport protein (MTP). A second pathway of triglyceride synthesis uses acylation of glycerol-3-phosphate to phosphatidic acid, dephosphorylation to diacylglycerol (DAG), and then acylation of DAG to TAG. A high-density, protein-rich particle synthesized in the enterocyte, containing apo B48 and apo AIV, is then packaged with the absorbed lipids to generate the chylomicron.22 This is then transported by vesicles to the basolateral membrane for exocytosis into the mesenteric lymph. The lipoproteins in the lymph enter the blood circulation via discharge from the thoracic duct.
Two ABCG half transporters, ABCG5 and G8, contribute to net cholesterol absorption in the gut through their ability to resecrete sterol that has been absorbed by the enterocyte back into the gut lumen, thereby reducing net sterol uptake.23 The total amount of diet-derived TAG that is delivered to the circulation varies and is influenced by multiple factors, including the fat content of the diet, the amount of phosphatidylcholine in the intestinal lumen, and the expression level of apo AIV in the enterocyte. Overall, humans have a remarkable ability to absorb large quantities of both cholesterol and fat; 35% to 60% of ingested cholesterol is absorbed, with the amount inversely correlated with hepatic cholesterol synthesis. Up to 600 g of fat is absorbed with 95% efficiency. In individuals with normal lipid metabolism, the ingested fat is cleared from the enterocyte in less than 14 hours, forming the basis for drawing serum lipids after a fast of this duration. Circulating chylomicrons are subsequently cleared from the plasma by the action of endothelial-bound lipoprotein lipase, which cleaves the TAG, enabling the liberated glycerol and free fatty acids to move into the adjacent adipose or muscle tissue. The resulting chylomicron remnant particle can be further metabolized by lipases and appears to be retained in the hepatic space of Disse, where it can bind to heparan sulfate proteoglycans and acquire additional apo E. The acquisition of the apo E enables the remnant to be cleared efficiently by either the LRP1 or LDL receptor, but patients with diabetes have accumulation of remnants and slower hepatic clearance.24,25
The other major triglyceride-rich lipoprotein, VLDL, is synthesized in the liver and contains apo B100 rather than apo B48 as its major structural protein.26 Like chylomicrons, VLDLs rely on MTP to combine lipids with the apo B protein to produce a nascent, lipid-poor lipoprotein.27,28 This VLDL can be secreted from the liver or further lipidated to produce a mature VLDL. Individuals lacking MTP activity develop abetalipoproteinemia, a disorder characterized by erythrocyte acanthocytosis, anemia, steatorrhea, spinocerebellar degeneration, and fat-soluble vitamin deficiency. Acquisition of lipid by the mature VLDL appears to depend on triglycerides that are stored in the cytosol. Increased fatty acid delivery to the liver, from dietary sources or that released by lipase activity in peripheral tissues, stimulates VLDL production. After secretion of the mature VLDL from the liver, its triglycerides are initially removed by the action of LPL, and the resulting intermediate-density lipoprotein (IDL) can be further catabolized by hepatic lipase to produce mature, cholesterol-rich LDL.
Low-Density Lipoprotein Metabolism
There is still some dispute about whether mature LDL can be secreted from the liver rather than always being derived from the progressive removal of triglyceride from VLDL/IDL precursors.29 The preponderance of data indicate that little if any LDL is directly secreted from the liver. Each LDL particle contains a single apo B molecule, which dictates that individuals heterozygous for apo B mutants will have two populations of LDL, one which carries a wild-type protein and one with a mutant protein. LDLs are utilized throughout the body as a source of exogenous cholesterol and are taken up by LDL receptors present at the plasma membrane. While LDL receptors are ubiquitous, the majority are expressed by the liver. The LDL receptor binds to both apo E– and apo B–containing lipoproteins. Because the affinity of the receptor is higher for apo E than for apo B, E-containing lipoproteins, such as IDL, can compete with LDL for uptake by the LDL receptor. However, there are other receptors that can bind apo E–containing lipoproteins, permitting their clearance but not that of LDL when LDL receptor activity is lost or diminished. Thus, mutations in the LDL receptor result in elevated LDL levels in the plasma, causing the autosomal co-dominant disorder, familial hypercholesterolemia, which is among the most common of Mendelian genetic disorders (see Genetics section later).30 The elucidation of the cholesterol homeostatic mechanisms controlled by the LDL receptor constituted one of the major advances in our understanding of human physiology and disease.31 The internalization of LDL by the LDL receptor leads to trafficking of the internalized lipoprotein to the endosome, where after fusion with lysosomes, the sterol in the lipoprotein is released and shuttled to the endoplasmic reticulum (ER). A sterol-sensing system in the ER regulates the movement of a membrane-bound transcription factor, sterol response element binding protein (SREBP), from the ER to the nucleus.32–37 SREBP works in concert with other transcriptional regulators to influence the expression of many of the cholesterol synthesis, metabolizing, and transport molecules, particularly in the liver. This elegant homeostatic system enables cells to reduce their de novo cholesterol synthesis when adequate sterol supplies are present and to increase synthesis and LDL receptor expression when cellular demands require more cholesterol.
High-Density Lipoprotein Metabolism
Although the relationship between elevated levels of HDL and lower rates of cardiovascular disease was recognized more than 50 years ago, the mechanism by which HDL protects against CHD is still only partially understood. HDL are initially produced as lipid-poor phospholipid discs containing apolipoprotein AI. Following interaction with the phospholipid/cholesterol transporter, apo AI, the particle acquires unesterified cholesterol from cellular stores.38 For macrophages in particular, this cholesterol efflux pathway appears to be critical to their ability to unload the excess cholesterol they acquire by phagocytosis of dead cells and by their unregulated uptake of lipoproteins by scavenger receptor pathways.14 Cholesterol is stored in an intracellular lipid pool, having been esterified at its 3′OH position by the activity of acyl coenzyme A (CoA):cholesterol O-acyltransferase (ACAT).39 A neutral cholesterol hydrolase cleaves the fatty acid from cholesterol ester to convert it back to free (unesterified) cholesterol when cellular signals call for the use or export of the sterol.40 This free cholesterol can be shuttled to the plasma membrane, where it can be transported by the action of ABCA1. The interaction of lipid-poor nascent HDL with ABCA1 appears to involve direct binding of apo AI to the transporter, but the actual lipid transfer process is poorly understood.41–43 Once the unesterified cholesterol transfers to the nascent HDL, it can be reesterified by the action of lecithin cholesterol acyltransferase (LCAT) and centralized in the lipid core of the growing HDL particle. This HDL particle can now interact with a second ABC transporter, ABCG1, and additional cholesterol can be acquired.44 The role of ABCA1 is well established, since loss of its activity leads to Tangier disease (see Genetics section) and the absence or near absence of HDL in the plasma. ABCG1’s precise function is still being elucidated. Once the HDL has acquired sufficient cholesterol and esterified it, it can donate this lipid back to lower-density lipoproteins in the plasma, such as VLDL, through the activity of cholesterol ester transfer protein (CETP). Alternatively, HDL can interact with a cellular receptor, SR-BI, to selectively transfer the cholesterol ester to cells, including the liver, where it could excreted in the bile as cholesterol or one of its bile acid derivatives.45 The protein component of HDL is cleared via the kidney, a process that appears to depend on the HDL being lipid depleted. The pathway described, moving cholesterol from cellular storage pools to HDL and ultimately to the liver, is termed the reverse cholesterol transport pathway. The activity of this pathway is postulated to explain at least in part the link between HDL levels and lower CHD risk. However, HDL also carries antioxidants and a host of proteins linked to complement and inflammation pathways that suggest it may be playing a more general role as a scavenger of molecules that stimulate inflammation. The complexity of its function has made targeting increases in HDL levels as a therapeutic objective challenging. Both animal and human data have established that not all methods of increasing HDL result in improvements in atherosclerosis.
Disorders of Lipid Metabolism in Patients With Diabetes
TYPE 1 DIABETES
Data dating back at least 30 years have documented that individuals with type 1 diabetes have increased rates of CHD.46–48 This risk has been estimated to be 10-fold or higher over that seen in age-matched controls without diabetes. In the Diabetes U.K. cohort of 23,751 subjects who were diagnosed with insulin-dependent diabetes prior to the age of 30, for example, those individuals 20 to 29 years of age had a standardized mortality ratio for ischemic heart disease of 11.8 for men and 44.8 for women. For those 30 to 39 years of age, the ratios were 8.0 and 41.6 for men and women, respectively.49 Despite the long and consistent reproducibility of this risk association, the pathophysiology underlying that relationship is still poorly understood. Unlike type 2 diabetes, where obesity and insulin resistance lead to changes in the standard lipid profile that are considered atherogenic, the type 1 diabetic who has not developed nephropathy and whose glucose is well controlled will typically have a normal serum lipid profile.50 In the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC), the mean LDL-C in the intensively treated group was 111 ± 29 mg/dL in women and 119 ± 31 in men; HDL-C levels averaged 63 ± 16 and 51 ± 14 for women and men, respectively. Serum triglyceride levels were also in the normal range, with women averaging 76 ± 37 mg/dL, and male values running moderately higher at 98 ± 67 mg/dL. These lipid values did not differ significantly from those of the patients enrolled in the conventional treatment group of the study. These findings make clear that the standard lipid profile provides few insights into the increased risk of CHD in type 1 diabetics.
More sophisticated assessments of serum lipoproteins, using nuclear magnetic resonance (NMR), can reveal differences in lipoprotein sizes that distinguish diabetic lipids from those of nondiabetics. A study of 194 diabetics from London, all diagnosed with type 1 diabetes before the age of 25 and not receiving any renal replacement therapies, demonstrated that LDL size and subclass distribution were similar between the men in both groups.51 In contrast, female diabetics had fewer large and more small LDL, producing an overall reduction in LDL size. However, both men and women diabetics had more large and fewer small HDL than did the nondiabetic cohort, yielding an increased overall HDL size. Whereas the LDL size change in women would be considered proatherosclerotic, the HDL differences are thought to be antiatherogenic. These findings led the authors to conclude that the pathogenic significance of the lipoprotein size differences was uncertain.51 The results of the London study appear to differ from those reported in a similar analysis done in the DCCT cohort, although the comparators used in the two studies make direct comparison of the two reports challenging. In the DCCT analysis, a nondiabetic control group was not included, so the authors compared the NMR analysis of lipoproteins in men to those in women. This approach found higher LDL particle concentrations and more small-sized particles in the men. HDL sized decreased, and fewer large HDL were also found in the men. Although the authors stated that both genders had abnormal levels of several of the NMR-classed lipoproteins, these judgments were based on National Cholesterol Education Program (NCEP) optimal levels rather than population normative values, so it is not clear that matched, nondiabetic cohorts would have differed greatly.50
Surprisingly, epidemiologic risk-factor assessment of the correlates of CHD in type 1 diabetes shows a weak association to the one metabolic risk that predominates in the type 1 population: hyperglycemia.52,53 It was therefore somewhat unexpected when a 17-year follow-up of the DCCT cohorts showed that the intensive treatment group had a 57% reduction in the risk of nonfatal myocardial infarction, stroke, or death from cardiovascular disease compared to the conventional treatment group (95% CI, 0.21 to 0.88; P = 0.02). Unfortunately, the DCCT/EDIC findings, if generalizable to the care of diabetics in the community at large, appear not to have translated yet into that community, since the cumulative incidence of CHD in type 1 diabetics has changed little in the past few decades.54 Unlike CHD, peripheral arterial disease,55 amputation,56 and stroke57 rates have all been reported to correlate with levels of hyperglycemia. Possible explanations for the differences between CHD and other forms of CVD in type 1 diabetes are discussed in further detail in Chapter 51.
TYPE 2 DIABETES
There are alterations in multiple lipoprotein metabolism pathways in type 2 diabetics that lead to the typical abnormal lipid profile commonly seen. This profile includes hypertriglyceridemia secondary to high VLDL levels, as well as increased amounts of sdLDL and lower levels of HDL cholesterol. LDL cholesterol levels are typically not higher in diabetic populations when compared to well-matched nondiabetic subjects. In an elderly Finish population, for example, approximately 30% of diabetic subjects had serum triglyceride levels greater than 200 mg/dL compared to 13% of nondiabetic subjects. HDL cholesterol levels were less than 35 mg/dL in about 25% of diabetics compared to 12% of nondiabetics. In this study, elevated LDL levels as defined by an LDL greater than 130 mg/dL were actually more prevalent in the nondiabetic population than in the diabetics.58,59
The causes of classic diabetic dyslipidemia are only partly understood. Individuals with insulin resistance and type 2 diabetes overproduce mature VLDL in the liver. This is accompanied by a slower clearance of triglyceride-rich lipoproteins via reduced activity of lipoprotein lipase. Coupled with decreased receptor uptake of the remnant and IDL particles that form after LPL partially depletes VLDL of its triglyceride, diabetes produces substantial elevations in serum triglyceride levels. Production of sdLDL appears to be tightly linked to the resulting hypertriglyceridemia insofar as cholesterol ester transfer protein (CETP) exchanges triglyceride from VLDL for cholesterol ester from LDL, producing a triglyceride-enriched LDL.60 This lipoprotein appears to be a preferred substrate for hepatic lipase, which hydrolyzes the triglyceride to produce an LDL that is smaller, lower in cholesterol ester content, and higher in density than LDL produced in the absence of excess VLDL. The diabetic with increased small dense LDL levels has a higher LDL particle number for an equivalent serum LDL cholesterol concentration than does an individual with normal-density LDL. This increased particle number, whether measured by nuclear magnetic resonance assays or serum apo B levels, is associated with an increased risk of CAD.12
Thus, the central metabolic change that accounts for much of the diabetic dyslipidemia is the overproduction of VLDL by the liver. Higher levels of circulating free fatty acids, arising from increased adipose deposits and insulin deficiency or resistance, contribute to higher storage levels of triglyceride in the liver. Increased fat content of the liver is in turn correlated with increased rates of lipidation and secretion of nascent VLDL. This VLDL appears to be normal in size, indicating that higher VLDL triglyceride levels derive from higher particle numbers rather than larger triglyceride-enriched lipoproteins. Administration of insulin reduces the concentration of free fatty acids in the circulation and also suppresses VLDL production by inhibiting the hepatic generation of the early apo B, lipid-poor particle. The molecular mechanisms accounting for VLDL production changes are not fully elucidated, but there is evidence that insulin activates a phosphatidylinositol-3 (PI-3) kinase pathway that inhibits apo B secretion while activating a mitogen-activated protein kinase (MAPK) that down-regulates MTP expression.61,62 Insulin effects on transcriptional regulators of VLDL secretion may also be involved. In vivo metabolic studies suggest that the prevalence of hypertriglyceridemia and low HDL cholesterol levels is higher in diabetics who are insulin resistant as opposed to insulin deficient, accounting for the higher prevalence of these disorders in type 2 versus type 1 diabetics.
Genetic Basis of Lipid Disorders
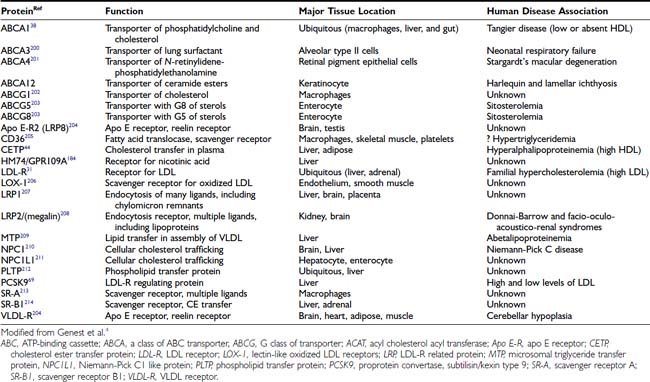
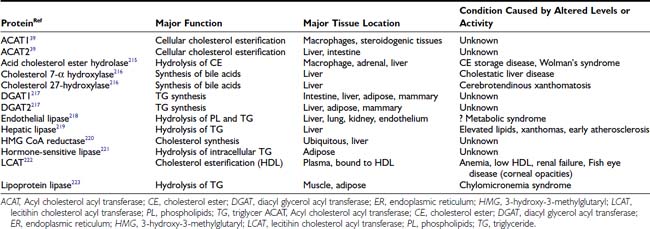
Although metabolic disorders associated with diabetes, hypothyroidism, and nephropathy can exacerbate or even cause hyperlipidemia (Table 42-4), many individuals have none of these problems. Individuals with the greatest deviation from normal levels of lipoproteins often have a single gene defect that is responsible for their lipid disorder. The vast majority of hyperlipidemic patients, however, do not have a monogenic defect. Rather, they are most likely to have a polygenic disorder and an additional contribution from environmental factors (e.g., excessive saturated fat intake for high LDL levels; obesity or smoking for lower HDL levels). The application of the NCEP treatment guidelines does not require that a genetic diagnosis be made, because treatment is based on phenotype (lipid levels) and not genotype. Nevertheless, the identification of the causes of monogenic lipid disorders has been critical to our understanding of both normal and abnormal lipoprotein physiology. The most important lipid disorders affecting coronary disease risk are those that increase the serum LDL level or reduce the HDL level, and these are briefly presented next.
Table 42-4. Secondary Causes of Hyperlipidemia and Dyslipidemias
| Metabolic/hormonal | Diabetes |
| Hypothyroidism | |
| Lipodystrophy | |
| Polycystic ovarian disease | |
| Hepatic | Primary biliary cirrhosis |
| Other forms of cirrhosis | |
| Renal | Chronic renal failure |
| Nephrotic syndrome | |
| Dietary | Alcohol |
| Foods highly enriched in cholesterol and saturated fat | |
| Medications | Estrogens |
| Glucocorticoids | |
| Anti-HIV treatments, especially protease inhibitors | |
| Oral androgens and anabolic steroids | |
| Thiazide diuretics | |
| Beta-blockers | |
| Retinoic acid | |
| Antipsychotics |
MONOGENIC LOW-DENSITY LIPOPROTEIN DISORDERS
The molecular basis of four monogenic disorders that primarily affect LDL levels have been characterized: (1) familial hypercholesterolemia, (2) familial defective apo B, (3) autosomal recessive hypercholesterolemia, and (4) PCSK9 mutations. These disorders illustrate distinct mechanisms leading to elevated LDL levels.
Familial Hypercholesterolemia
Familial hypercholesterolemia (FH) is one of the most thoroughly studied and common genetic disorders of mankind. Approximately 1 in 500 individuals carry a mutation in one allele encoding the LDL receptor. These mutations results in defective clearance of LDL from the blood and a rise in serum total and LDL cholesterol levels. The elucidation of this defect and its associated cell biology led to insights into the homeostatic control of cholesterol metabolism that transformed the lipid field.31 Several hundred individual mutations in the LDL receptor have now been identified. The gene, located on chromosome 19 and spanning 45 kb, has 18 exons that encode a mature protein of 839 amino acids. The inheritance of the disorder is autosomal co-dominant. Heterozygous patients have LDL cholesterol levels in the 200 to 500 mg/dL range, and the rare homozygous FH patient typically has an LDL cholesterol above 500 mg/dL. Heterozygous patients commonly have tendon xanthomas and premature CAD, whereas these are universal in the untreated homozygous individual. Mutations in the LDL receptor have been described that affect multiple aspects of receptor biology, ranging from defective LDL binding to abnormal trafficking of the receptor to the plasma membrane. The effect of all of these mutations is to reduce LDL and IDL clearance from the blood. The latter effect results in an enhanced conversion of IDL to LDL. The net result is markedly elevated LDL cholesterol levels in the plasma and a strong predisposition to early CHD if the disorder is not treated. FH heterozygotes typically respond well to 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (statins), though additional therapies may be needed to reach desirable LDL levels. FH homozygotes respond poorly to reductase inhibitors and inevitably develop early atherosclerotic vascular disease if LDL apheresis or liver transplantation is not performed.
Familial Defective Apolipoprotein B
Familial defective apo B (FDB) is an autosomal co-dominant disorder caused by mutations near the carboxy terminus of apo B.63,64 Apo B is a 4536-amino-acid protein encoded by a gene on chromosome 2 that is composed of 29 exons. The most commonly identified mutations in FDB affect an arginine at position 3500 that is either mutated to a glutamine or tryptophan. Most individuals with FDB are heterozygous for their mutation, and there is only one molecule of apo B per LDL particle, so the LDL in the serum of these individuals is a mixture of normal LDL and FDB LDL. The LDL containing the defective apo B does not bind normally to the LDL receptor and is therefore cleared more slowly. The explanation for the lower receptor affinity of LDL carrying the FDB mutations is not known with certainty. There is evidence, however, of a molecular interaction between the arginine at position 3500 and tryptophan at 4369 that appears to be necessary for the adoption of an apo B conformation recognized by the LDL receptor. Patients with FDB have elevations in their LDL levels that fall within the range of those seen in FH heterozygotes, but the mean is generally lower than the mean in FH cohorts. There is considerable variability in both genetic populations; however, making generalizations about the diagnostic value of this measurement not very useful. Tendon xanthomas are common but not universal, and premature CAD is prevalent. Treatment with reductase inhibitors or other standard LDL-lowering drugs is typically quite effective in FDB patients.
Autosomal Recessive Hypercholesterolemia
Studies of families in which LDL levels were inherited in an autosomal recessive pattern led to a successful genetic linkage analysis that mapped the genetic defect in these families to the short arm of chromosome 1. Candidate gene sequencing led to the identification of mutations in a gene now designated autosomal recessive hypercholesterolemia (ARH) and comprising nine exons and eight introns. Patients diagnosed with ARH to date have been clinically similar to patients who are homozygous for LDL receptor mutations in that they have markedly elevated LDL cholesterol levels (typically > 500 mg/dL), tendon xanthomas, and premature coronary atherosclerosis.65 The autosomal recessive mode of inheritance is an important differentiating factor between ARH and familial hypercholesterolemia, as is greater LDL receptor activity, measured in cultured fibroblasts taken from the patients. The defect in ARH patients appears to affect liver cholesterol metabolism disproportionately, and current data indicate that the ARH gene product likely serves as an adaptor protein required for LDL receptor internalization via clathrin-coated pits.66,67 ARH patients do respond to HMG CoA reductase inhibitors, but this treatment is usually inadequate to control their markedly elevated LDL cholesterol levels, making them candidates for LDL apheresis.
PCSK9 Mutations
Proprotein convertase subtilisin kexin 9 (PCSK9) is a 72-kD protease that is highly expressed in the liver and regulates the levels of LDL receptors acting at the plasma membrane.68,69 Gain-of-functions mutations in PCSK9 result in lower LDL receptor levels, whereas loss-of function mutations increase those levels. Serum LDL levels are inversely related to liver LDL receptor expression; the activity of the latter clears the former from the circulation. Thus, gain-of-function mutations are associated with higher serum LDL levels and increased risk of CHD, and loss-of-function mutations result in lower LDL levels and decreased CHD risk. The role of PCSK9 in lipid metabolism was originally recognized when it was discovered that specific heterozygous mutations in the protein were associated with autosomal dominant hypercholesterolemia. Approximately 1 in 50 African Americans has one of two nonsense mutations in PCSK9, causing LDL cholesterol levels to fall approximately 30%. Caucasians can carry a missense mutation that reduces LDL cholesterol by approximately 15%. Individuals with complete loss of PCSK9 activity have extraordinarily low levels of LDL-C, but appear to be otherwise normal. The mechanism by which PCSK9 regulates LDL receptor activity is still being elucidated, but it appears that the protease can bind to the receptor, targeting it for degradation in the lysosome. This action does not depend on the protein’s enzymatic activity, and exogenously administered PCSK9 can reduce LDL receptor expression and serum LDL-C levels. The mechanism by which PCSK9 works makes it an extremely attractive target for pharmaceutical intervention; therapeutics that block PCSK9 function are being actively pursued.
MONOGENIC HIGH-DENSITY LIPOPROTEIN DISORDERS
Three distinct monogenic disorders cause markedly reduced levels of HDL in the plasma by different mechanisms. They are (1) Tangier disease, (2) LCAT deficiency, and (3) apo AI mutations.
Tangier Disease
Tangier disease was first recognized in 1960 in a sibling pair living on Tangier Island in the Chesapeake Bay in Virginia. Enlarged, yellow-orange tonsils and little or no circulating HDL cholesterol are the classic findings in the disorder. Subsequently, cases have been identified in which the presenting symptom was a peripheral neuropathy. Patients may also have hepatosplenomegaly. Serum LDL and total cholesterol levels are usually quite low, while serum triglyceride values are moderately elevated. Tangier disease is an autosomal recessive disorder. The cause of Tangier disease was identified by several groups, following mapping of the gene defect to chromosome 9. Candidate gene analysis in the appropriate genetic interval identified mutations in an ATP-binding cassette (ABC) transporter as the cause. The transporter, now called ABCA1 (ATP-binding cassette, subfamily A, member 1), is a full-length ABC transporter transmembrane protein that is predicted to span the plasma membrane 12 times. ABCA1 is a 2261-amino-acid protein encoded by a gene spanning 50 exons. Approximately 50 mutations have been identified to date in the gene.70 ABCA1 mediates the efflux of cholesterol from cholesterol-enriched cells when stimulated by the major apoprotein of HDL, apo AI.71 This activity is lost in Tangier patients and is reduced by approximately half in carriers of one abnormal ABCA1 allele. The mechanism of the movement of cholesterol from inside the cell to outside the cell has not been established. Individuals with Tangier disease and heterozygous carriers of ABCA1 mutations (a disorder called familial hypoalphalipoproteinemia [FHA]) both appear to have increased risk of premature coronary disease. There is no specific therapy for this disorder.
Lecithin Cholesterol Acyltransferase Deficiency
The esterification of free cholesterol in circulating lipoproteins is catalyzed by a plasma enzyme called lecithin cholesterol acyltransferase. Two clinically separable syndromes result from a deficiency of LCAT. Fish eye disease is due to a partial deficiency of LCAT, with patients presenting with dense corneal opacities and very low HDL cholesterol levels. Familial LCAT deficiency arises from a nearly completely absence of LCAT activity and produces a more severe syndrome characterized by corneal opacities, anemia, and proteinuric renal failure.72 The serum lipid and lipoprotein profile in the more severe disorder is characterized by normal or increased triglyceride levels, reduced LDL cholesterol values, and markedly diminished HDL levels. The gene encoding LCAT is located on chromosome 16 and is composed of 6 exons. Cleavage of a signal peptide of 24 amino acids converts the proenzyme from a 440-amino-acid precursor to the final 416-amino-acid glycoprotein that circulates in the plasma. When unesterified cholesterol from tissues is transferred to HDL, either by passive diffusion or by ABCA1-mediated lipid transport, LCAT’s activity esterifies the transferred cholesterol, trapping it in the HDL core. Since cholesterol ester is more hydrophobic than unesterified cholesterol, it is energetically unfavorable for the cholesterol ester to transfer back to the cell of origin. These steps of cholesterol transfer and esterification are the initial events in the reverse cholesterol transport pathway whereby cholesterol is moved from peripheral tissues back to the liver. Apo AI is the major activator of LCAT activity, accounting for the predominant effect of the enzyme deficiency on HDL levels. LCAT does, however, contribute to esterification of cholesterol in lower-density lipoproteins as well. Despite very low HDL levels, patients with either fish eye disease or familial LCAT deficiency do not seem to have a predilection for very early coronary atherosclerosis. The small number of patients with the disease, some of whom have been found to have CHD, make it difficult to determine if the risk of coronary atherosclerosis is substantially altered by the enzyme deficiency. There is no specific treatment for LCAT deficiency. Corneal and kidney transplantation are performed in these patients in order to ameliorate their major clinical disabilities.
Apolipoprotein AI Mutations
Apo AI is the major structural protein of HDL. The gene encoding apo AI is located on chromosome 11 and comprises 4 exons. Following cleavage of the signal and prohormone sequences, a 243-amino-acid mature protein is produced. The protein has multiple repeats of an amphipathic helical structure that enables it to interact with both lipid and aqueous environments. Mutations that cause profound alterations in apo AI structure or expression have been reported, though they are extremely rare.73–76 The individuals carrying these mutations have virtually no circulating HDL and typically develop early CHD. There are, however, a number of case reports of apo AI mutations that are not associated with early CHD. Corneal opacities and xanthomas have been documented in many but not all individuals with major apo AI mutations. Most patients harboring these mutations have been found to be homozygous for the gene defect, usually as a result of consanguinity. Heterozygotes carrying these mutations commonly have half-normal HDL cholesterol levels, although cases with greater reductions in HDL levels have been reported, suggesting that some mutations may exert a dominant-negative effect. HDL typically contains four apo A molecules per particle (either four apo AI or two apo AI and two apo AII proteins), so a heterozygous individual carrying an expressed apo AI mutant would have at least one mutant apo AI on most HDL particles. The atherosclerosis of individuals with structural mutations in apo AI appears to be much more pronounced than that seen in patients with either LCAT deficiency or Tangier disease. No specific therapy is available for this disorder, but aggressive LDL lipid-lowering therapy is justified.
Diagnosis of Lipid Disorders
The diagnosis of a lipid disorder should be based on more than one measurement of serum lipids, because combined analytic and biological variations in serum lipids range from 10% to 20%. The technology for measuring LDL levels directly has improved steadily over the years, but in most laboratories, it remains a calculated value. To perform this calculation, the total and HDL cholesterol levels as well as the triglyceride value are measured. The LDL cholesterol concentration is then estimated using the following formula devised by Friedewald et al.:

The triglyceride/5 factor represents an estimate of VLDL cholesterol. The validity of this formula for estimating LDL cholesterol has been confirmed by ultracentrifugal measurement of lipoprotein levels and remains reasonably accurate as long as the total triglyceride is less than 400 mg/dL. In order to obtain an accurate calculation, patients must fast for at least 12 hours to clear their blood of any chylomicrons; these lipoproteins distort the triglyceride ratio on which the Friedewald formula relies. If the triglyceride level is greater than 400 mg/dL, the LDL cholesterol must be determined by alternative methods. Apo B100, which is present in both LDL and VLDL can be measured directly to get an assessment of lower-density lipoprotein particle numbers, but this is not yet routinely done in the United States. With increasing evidence for a greater atherogenicity of smaller, denser LDL particles, more sophisticated assessments of LDL number and composition are being introduced into clinical practice, such as nuclear magnetic resonance spectroscopy. The place of these more sophisticated and expensive assays in the routine diagnostic evaluation of most patients remains unsettled. It appears that most patients at high risk for CHD can still be identified using more traditional laboratory markers.
Before embarking on a treatment plan in a patient with hyperlipidemia, one must exclude other medical conditions that cause lipids to rise as a secondary consequence. The most common clinical conditions that cause this to occur are obesity, diabetes, and hypothyroidism. The latter two are best screened using a serum glucose (or hemoglobin [Hb] A1c) measurement and a thyrotropin-stimulating hormone (TSH) level, respectively. Many medications commonly cause a secondary hyperlipidemia, with antiretroviral therapy, estrogens, and glucocorticoids heading the list.
Increasingly, the diagnosis of severe hyperlipidemic syndromes due to rare mutations in key proteins of the lipid metabolism system can be made by genetic testing. Using buccal smears or circulating white blood cells present in a blood sample, DNA can be extracted and mutations easily detected. At the present time, these assays are used to guide family counseling or provide interested patients insights into the cause of their disorder, but the results rarely affect clinical decision making or therapeutic choices. Most of these assays remain research tools, with few being covered by medical insurance. The role of genetic diagnostics in medicine is in rapid flux, and new discoveries may change this situation in the next few years.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


