Total Body Iron
Iron endowment varies with age and sex. Full-term infants begin life with approximately 75 mg/kg body weight of iron, primarily acquired from their mothers during the third trimester of gestation. These abundant stores are rapidly depleted over the first few months of life, and most young children have tenuous iron balance, as their intake must keep pace with rapid growth. Requirements decrease after adolescence, and men have a small gradual increase in iron stores throughout life. The body iron content of normal adult men is 50 mg/kg body weight or greater. In contrast, postpubertal women have increased losses of iron due to menstruation, pregnancy, and childbirth, resulting in a body iron endowment averaging 35 mg/kg. After menopause, women accumulate iron linearly in parallel with adult men.
Most of the body iron is found in heme-containing oxygen transport and storage proteins, including hemoglobin and myoglobin (
Table 23.1). Smaller amounts are incorporated into enzymes with active sites containing heme or iron-sulfur clusters, including enzymes of electron transport chain, peroxidases, catalases, and ribonucleotide reductase. Most nonheme iron (approximately 1 g in adult men) is stored as ferritin or hemosiderin in macrophages
and hepatocytes. Only a tiny fraction of iron (˜0.1%) is in transit in the plasma, bound to the carrier protein, transferrin.
Iron Balance
Iron is not actively excreted from the body; it is eliminated only through the loss of epithelial cells from the gastrointestinal tract, epidermal cells of the skin, and, in menstruating women, red blood cells. On the basis of long-term studies of body iron turnover, the total average daily loss of iron has been estimated at ˜1 to 2 mg in normal adult men and nonmenstruating women.
6 Although iron is a physiologic component of sweat, only a tiny amount of iron (22.5
µg/L) is lost by this route.
7 Urinary iron excretion amounts to <0.05 mg/day and is largely accounted for by sloughed cells. Menstruating women lose an additional, highly variable amount over each menstrual cycle, from 0.006 (average) to more than 0.025 mg/kg/day.
8These iron losses are normally balanced by an equivalent amount of iron absorbed from the diet (1 to 2 mg/day). The bioavailability of iron in the U.S. diet has been estimated to be ˜16.6%, but only a fraction of dietary iron is absorbed and the amount of bioavailable iron is lower in many parts of the world. Fractional absorption of dietary iron can increase up to three- to fivefold (3 to 5 mg/day) if iron stores are depleted. Thus, iron balance is primarily, if not exclusively, achieved by control of absorption rather than by control of excretion.
Intestinal Absorption
Iron is absorbed in the duodenum, and humans and other omnivorous mammals have at least two distinct pathways for iron absorption: one for uptake of heme iron and another for ferrous (Fe
2+) iron. Heme iron is derived from hemoglobin, myoglobin, and other heme proteins in foods of animal origin, representing approximately 10% to 15% of iron content in the typical Western diet,
9 although heme-derived iron accounts for 2/3 of absorbed iron in meat-eating humans. Exposure to acid and proteases present in gastric juices frees the heme from its apoprotein. Heme is taken up by mucosal cells, but the specific receptor is still unknown.
10 Once heme iron has entered the cell, the porphyrin ring is enzymatically cleaved by heme oxygenase.
11 The liberated iron then probably follows the same pathways as those used by nonheme iron. A small proportion of the heme iron may pass into the plasma intact via heme exporter protein FLVCR (feline leukemia virus, subgroup C receptor),
12 which transfers heme onto a heme-binding protein, hemopexin.
13 Absorption of heme iron is relatively unaffected by the overall composition of the diet.
9Dietary nonheme iron is largely in the form of ferric hydroxide or loosely bound to organic molecules such as phytates, oxalate, sugars, citrate, lactate, and amino acids. Low gastric pH is thought be important for the solubility of inorganic iron. Dietary constituents may also have profound effects on the absorption of nonheme iron, making the bioavailability of food iron highly variable.
9 Ascorbate, animal tissues, keto sugars, organic acids, and amino acids enhance inorganic iron absorption, whereas phytates, polyphenols, and calcium inhibit it.
14 Depending on various combinations of enhancing and inhibitory factors, dietary iron assimilation can vary as much as tenfold.
Molecular Mechanisms of Iron Absorption
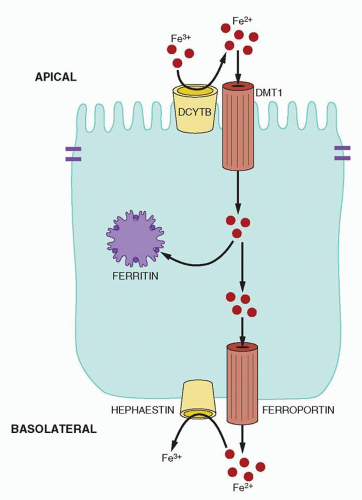
Absorption of nonheme iron occurs in the duodenum, where ferrous iron is imported into enterocytes by divalent metal ion transporter 1 (DMT1, also known as Nramp2, DCT1, SLC11A2).
15,16 DMT1 is a protein with 12 predicted transmembrane segments, which is expressed on the apical surface of absorptive enterocytes. Levels of DMT1 are markedly increased in iron-deficient animals
17 and in some animals with increased intestinal iron absorption resulting from other causes.
18 In addition to iron, DMT1 may also contribute to the absorption of other nutritionally important metals (e.g., Mn
2+).
19 Evidence in vitro demonstrates that DMT1 is a proton-coupled symporter.
15,20 The Na
+/H
+ exchanger in the duodenum is thought to create the “acid microclimate” that provides the proton electrochemical gradient as the driving force for iron uptake.
16DMT1 does not transport the Fe
3+ form of iron, but as discussed previously, most dietary nonheme iron arrives at the brush border as Fe
3+ ion. A duodenal cytochrome b-like ferrireductase enzyme, Dcytb (also called CYBRD1), likely reduces dietary Fe
3+ to make it a substrate for transport by DMT1.
21 Dcytb is induced in response to stimuli that increase iron absorption; however, since knockout mice appear to have normal metabolism, Dcytb may not be the only ferrireductase enzyme involved in absorption of nonheme iron.
22A variable fraction of iron taken into the mucosal cell is delivered to the plasma. The remainder is used by the cell or incorporated into ferritin,
23 an intracellular iron storage protein discussed in a later section. Iron retained in mucosal ferritin is not absorbed, rather, it is lost from the body when the senescent mucosal cells are sloughed into the intestine at the end of their 3- to 4-day lifespan (10
11 cells/day).
24Iron not stored in the absorptive enterocytes is transferred across the basolateral membrane of the cell to the lamina propria and ultimately to the plasma. This is accomplished, at least in part, by the iron exporter ferroportin (also known as FPN1, IREG1, MTP1, SLC40A1), a multi-transmembrane segment protein expressed on the basolateral surface of enterocytes.
25,26,27,28 Ferroportin is also expressed in other tissues involved in handling large iron fluxes including macrophages recycling iron from old red blood cells, hepatocytes storing iron, and placental trophoblast delivering iron from mother to fetus. Complete ablation of ferroportin in mice, including extraembryonic tissues, resulted in embryonic death,
28 whereas selective inactivation that preserved placental ferroportin expression resulted in live births, demonstrating the essential function of ferroportin in materno-fetal iron transfer. Ferroportin is also essential for systemic iron homeostasis as inactivation of ferroportin in all tissues in mice other than placenta led to the development of severe anemia resembling iron deficiency, and stemmed from the inability to mobilize iron from enterocytes, macrophages, and hepatocytes.
28Similar to apical iron uptake, basolateral iron efflux is aided by an enzyme that changes the oxidation state of iron. Ferroportin exports Fe
2+ and this iron must be oxidized to its Fe
3+ form to bind to transferrin. A membrane-bound multicopper oxidase, hephaestin, has been implicated in this process.
29 Hephaestin closely resembles ceruloplasmin,
30 which has been shown to be involved in efflux of iron from hepatocytes and macrophages. Mice carrying a large deletion in the X-chromosomal hephaestin gene
29 have impaired placental iron transfer and decreased intestinal iron absorption.
31,32 Their iron deficiency results in anemia during the neonatal period, but affected animals generally recover and show only subtle evidence of iron deficiency as adults. The simplest interpretation is that hephaestin is important in placental iron transport but dispensable for intestinal iron uptake. The functional relationship between hephaestin and ferroportin has not yet been defined in detail. A model incorporating current knowledge of nonheme iron transport is shown in
Figure 23.1.
Hepcidin and Systemic Regulation of Iron Metabolism
Hormone hepcidin has emerged as the key systemic regulator of iron homeostasis. Hepcidin is a small peptide of 25 amino acids, produced in the liver, secreted into the plasma, and excreted through the kidneys.
34,35 Hepcidin is structurally similar to antimicrobial peptides involved in innate immunity, but its main role is to inhibit iron fluxes into plasma.
36 Mutations in the human hepcidin gene cause severe, early-onset iron overload.
37 Mice lacking hepcidin also develop severe iron overload,
38 whereas transgenic mice constitutively expressing hepcidin have severe iron deficiency anemia.
39 A single injection of synthetic hepcidin into mice exerts a prolonged hypoferremic effect.
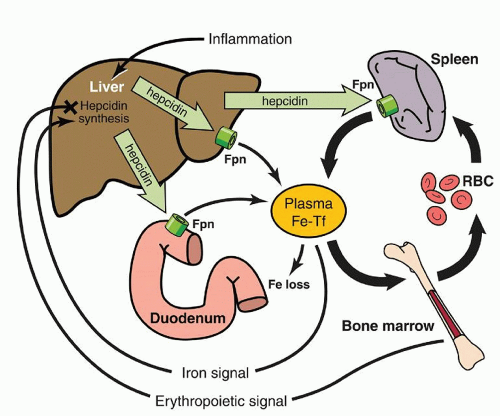
40 All of these effects can be explained by the biologic activity of hepcidin. Hepcidin binds to the iron exporter ferroportin, causing ferroportin endocytosis and lysosomal degradation,
41 and resulting in cessation of cellular iron export. Thus, hepcidin directly and coordinately controls the entry of iron into the plasma from ferroportin-expressing cells, including absorptive cells of the intestine and tissue macrophages (
Fig. 23.2).
Alterations in hepcidin production lead to changes in iron absorption and recycling. Multiple signals are known to regulate hepcidin expression, including iron, erythropoiesis, inflammation, and growth factors.
As with other hormones that are feedback regulated by the substances whose concentration they control, hepcidin expression is controlled by iron. When iron is deficient, hepatocytes produce less hepcidin, allowing more iron to enter plasma. When iron is abundant, hepcidin production increases, limiting further iron absorption and release from stores. In human volunteers ingesting a single dose of oral iron, hepcidin concentrations in urine increased within several hours and were proportional to the increase in transferrin saturation.
42 Serum hepcidin concentrations in healthy subjects also correlate with body iron stores as reflected by serum ferritin.
43 The specific forms of iron that increase hepcidin synthesis may include diferric plasma transferrin and intracellular iron in hepatocytes, and the transduction pathways that regulate hepcidin synthesis in response to these iron forms are just now being elucidated.
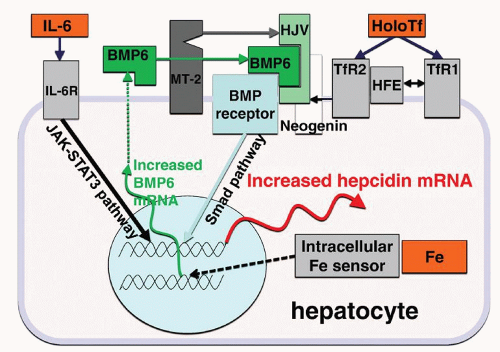
The two transferrin receptors TfR1 and TfR2, and HFE, an MHC class I-like membrane protein, may serve as holotransferrin sensors.
36 HFE can interact with both transferrin receptors, but this interaction is modulated by holotransferrin concentrations. Because HFE and holotransferrin binding sites on TfR1 overlap, increasing concentrations of holotransferrin result in displacement of HFE from TfR1, and free HFE then interacts with
TfR2.
44,45 TfR2 protein is further stabilized by binding of holotransferrin.
46 The holotransferrin/HFE/TfR2 complex then stimulates hepcidin expression through an incompletely understood pathway, possibly by potentiating BMP pathway signaling. HFE, however, may also regulate hepcidin expression without complexing with TfR2.
47 The role of HFE or TfR2 in hepcidin regulation by iron is supported by genetic evidence: HFE and TfR2 mutations in humans or mice cause hepcidin deficiency and an adult form of hemochromatosis.
36The BMP pathway with its canonical signaling via Smad proteins has a central role in the regulation of hepcidin transcription. BMP receptors are tetramers of serine/threonine kinase receptors, with two type I and two type II subunits. Recent data indicate that type I subunits Alk2 and Alk3 and type II subunit ActRIIA and BMPRII are specific BMP receptors involved in iron regulation.
48,49 In the liver, BMP pathway signaling to hepcidin is modulated by a coreceptor hemojuvelin and, at least in mice, by the ligand BMP6.
50,51,52 Loss of hemojuvelin or BMP6 in mice decreases hepcidin expression and impairs hepcidin response to acute or chronic iron loading.
53 In humans, hemojuvelin mutations result in severe hepcidin deficiency and cause juvenile hemochromatosis.
54 No pathogenic human mutations in BMP6 have yet been identified. It remains to be clarified how BMP receptors and hemojuvelin interact with iron-sensing molecules that regulate hepcidin expression.
Two other membrane proteins, a serine protease matriptase-2 (MT-2, also called transmembrane protease serine 6, TMPRSS6) and the receptor neogenin also influence hepcidin synthesis, likely by modulating hemojuvelin concentration on the cell membrane.
55,56 Whether these molecules are involved in iron sensing is unclear, but the concentration of hepatic MT-2 is acutely increased by iron deficiency.
57 Mutations in MT-2 in humans and in mice lead to increased hepcidin expression and development of iron-restricted anemia, discussed later in this chapter.
As already mentioned, hepcidin synthesis also increases in response to stored intracellular iron in hepatocytes but the underlying mechanism is yet unclear. Hepatic BMP6 mRNA increases in response to iron loading, suggesting that BMP6 may be a mediator of intracellular iron signal.
58 However, hepcidin increase after chronic iron loading is still observed in BMP6-deficient mice,
53 indicating that additional pathways must play a role in hepcidin regulation by intracellular iron.
Figure 23.3 summarizes our current understanding of the molecular mechanisms involved in hepcidin regulation by iron.
Hepcidin synthesis, and consequently absorption of iron and its availability for erythropoiesis, is also regulated by erythropoiesis itself. Increased erythropoietic activity due to bleeding, hemolysis, or administration of erythropoietin in humans or mice causes hepcidin suppression.
59,60,61 Very low hepcidin concentrations are observed in patients with absolute iron deficiency anemia, or anemias with high erythropoietic activity.
43 In turn, low hepcidin allows increased absorption of dietary iron and release of iron from stores, thus increasing iron supply to the bone marrow for hemoglobin synthesis. The mechanisms by which erythropoiesis regulates hepcidin production are not well understood, but it is thought that erythroid precursors in the bone marrow secrete a factor which exerts its effect on hepatocytes and causes hepcidin suppression.
60The suppressive effect of erythropoiesis on hepcidin is particularly prominent in diseases with ineffective erythropoiesis such as
β-thalassemia,
62,63 where erythrocyte precursors massively expand but undergo apoptosis rather than mature into erythrocytes. The severe suppression of hepcidin, particularly in untransfused
β-thalassemia, leads to increased iron absorption and the development of lethal iron overload. Growth differentiation factor GDF-15, a BMP-related protein released by erythroid precursors during cellular stress or apoptosis, may contribute to hepcidin suppression in anemias with expanded but ineffective erythropoiesis.
64 However, GDF-15 does not seem to be involved in homeostatic hepcidin suppression by effective erythropoiesis.
65Hepcidin gene expression is induced in response to inflammatory cytokines,
66 accounting for iron sequestration in the anemia of inflammation (
see Chapter 45).
59,67 IL-6 signaling through the STAT-3 pathway seems to be a key regulator of hepcidin expression in inflammation.
68,69 Apart from increased transcription, elevated hepcidin concentrations in circulation could result from impaired renal clearance of hepcidin. Because of its small size (2.7 kDa), hepcidin is filtered through the glomerular membrane and is then taken up and degraded in the proximal tubule. A fraction of the filtered hepcidin is excreted into urine, and urinary hepcidin concentrations are proportionate to plasma hepcidin levels in healthy subjects.
43 In chronic kidney diseases, however, hepcidin clearance is impaired,
70 contributing to the hormone accumulation in plasma.
Regulation of Iron Absorption by Intracellular Mechanisms
In addition to the regulation by systemic signals, iron absorption is subject to local regulation by intracellular mechanisms in duodenal enterocytes. At least two mechanisms have been described: one related to the enterocyte iron levels and the other to the hypoxia pathway.
Intracellular iron levels in enterocytes (and most other cells) are sensed by two iron-regulatory proteins (IRP1 and IRP2).
71 When cytoplasmic iron is low, IRPs bind to iron-regulatory elements (IREs),
72 stem-loop structures located in the 5′ or 3′ untranslated regions of different mRNAs. Binding of IRPs to 3′ IREs stabilizes mRNAs by protecting them from endonucleolytic cleavage, resulting in increased protein synthesis. mRNAs which contain 3′ IREs encode iron uptake proteins such as transferrin
receptor and a DMT1 isoform. In contrast, IRP binding to 5′ IREs blocks translation of mRNA, by preventing the interaction with the cap-binding complex eIF4F and the small ribosomal subunit,
73 resulting in decreased protein synthesis. 5′ IREs are present in ferritin mRNA and one of ferroportin transcripts. In iron-replete cells, neither IRP binds to IREs, resulting in greater degradation of 3′ IRE-containing transcripts (e.g., TfR1), and increased translation of 5′ IRE-containing transcripts (e.g., ferritin), thus shifting the cellular phenotype from an iron-importing to an iron-storing one.
How does intracellular iron concentration modulate IRP binding to IREs? IRP1 contains a 4Fe•4S iron-sulfur cluster that, when saturated with iron, converts IRP1 to a cytosolic aconitase that catalyzes the conversion of citrate to isocitrate.
74,75,76 In this enzyme form, IRP1 has low affinity for IREs in mRNAs. When iron-poor, IRP1 loses its aconitase activity and greatly increases its affinity for IREs. IRP2 is less abundant than IRP1 and does not have an identified enzyme function. Instead, intracellular iron levels control its degradation.
77,78 In iron-replete cells, IRP2 is ubiquitinated by an iron-sensitive E3 ubiquitin ligase complex containing FBXL5 protein,
79,80 and ubiquitinated IRP2 is degraded by proteasomes.
Targeted inactivation of the genes encoding murine IRPs has shed light on the roles of IRP1 and IRP2 in vivo. It is still not entirely clear why it is necessary to have two IRPs, but they may respond differently over the physiologically relevant range of oxygen tensions.
81 There are no obvious consequences from the loss of IRP1 alone, and loss of IRP2 results in mildly abnormal iron homeostasis and iron-deficient erythropoiesis.
82,83 Loss of both IRP proteins is incompatible with life.
84 Tissue-specific knockout of both IRPs in the intestine of mice confirmed their role in the regulation of iron transport in enterocytes. These animals had higher protein levels of ferroportin and ferritin and lower protein levels of DMT1. The knockout mice also had impaired intestinal function and died within 4 weeks of birth.
85Even though IRPs regulate translation of ferroportin mRNA through its 5′ IRE, duodenal ferroportin protein expression is paradoxically increased in iron deficiency. Recently, an alternative ferroportin transcript (FPN1B) was identified that could explain this phenomenon. FPN1B, which is highly expressed in the duodenum, lacks IRE and thus is not subject to translational repression by IRPs when intracellular levels are low.
86 This ensures that the transfer of iron from absorptive enterocytes into plasma continues even when those cells are iron deficient.
Dcytb mRNA also does not contain an IRE but is strongly upregulated in iron-deficient duodenum,
87 indicating additional regulation of transcription of iron-related genes. Hypoxia-inducible factor (HIF) transcription factors may serve as important local regulators of intestinal iron absorption. Iron deficiency induced HIF signaling in duodenum of mice, and caused increased DcytB and DMT1 expression, and an increase in iron uptake.
88 Accordingly, targeted deletion of Hif-2
α in the intestine resulted in dramatic decrease in the expressions of DMT1-IRE and Dcytb mRNA.
89 In vitro studies further demonstrated that HIF-2
α directly binds to the promoters of DMT1 and Dcytb, activating their transcription. Thus, HIF-2
α appears to be one of the critical local regulators of iron absorption.
Plasma Transport
The plasma iron-binding protein, transferrin, is a glycoprotein with a molecular weight of approximately 80 kDa.
90 Transferrin is synthesized chiefly in the liver and actively secreted by hepatocytes, but lesser amounts are made in other tissues, including the central nervous system, the ovary, the testis, and helper T lymphocytes (T4
+ subset). The rate of synthesis shows an inverse relationship to iron in stores; when iron stores are depleted, more transferrin is synthesized, and when iron stores are overfilled, the level of transferrin decreases. Transferrin keeps iron nonreactive in the circulation and extravascular fluid, delivering it to cells bearing transferrin receptors.
Transferrin can be measured directly using immunologic techniques; and normal concentration in the plasma is approximately 2 to 3 g/L. Alternatively, transferrin is quantified in terms of the amount of iron it will bind, a measure called the total iron-binding capacity (TIBC; normal values for plasma iron and TIBC are given in
Appendix A). In the average subject, the plasma iron concentration is ˜18
µmol/L (100
µg/dl), and the TIBC is ˜56
µmol/L (300
µg/dl). Thus, only about one third of the available transferrin binding sites are occupied, leaving a large capacity to deal with excess iron. Plasma iron concentration varies over the course of the day, with the highest values in the morning and the lowest in the evening. Levels of serum transferrin are more constant, and there is no apparent diurnal variation in TIBC. General practice has been to evaluate transferrin saturation using a first morning, fasting sample to standardize the results, but this may not be helpful.
91Transferrin has two homologous iron-binding domains, each of which binds an atom of trivalent (ferric) iron.
92 The iron atoms
are incorporated one at a time and appear to bind randomly at either or both of the two sites. When binding is complete, the iron lies in a pocket formed by two polypeptide loops. One mole of anion, usually carbonate or bicarbonate, is taken up, and 3 moles of hydrogen ion are released for each mole of iron bound. There are functional differences between the two iron-binding sites,
93 but it is not clear that these have physiologic importance.
Under physiologic circumstances, ferric iron binds to transferrin with very high affinity, with an affinity constant of ˜1-6 × 10
22 M
-l. The affinity of iron-transferrin interaction is pH-dependent, decreasing as pH is lowered. Other transition metals, such as copper, chromium, manganese, gallium, aluminum, indium, and cobalt, can be bound by transferrin but with less affinity than iron.
Iron Delivery to Erythroid Precursors
The biologic importance of transferrin in erythropoiesis is illustrated by abnormalities observed in patients and mice with congenital atransferrinemia.
94,95 and 96,97,98 When transferrin is severely deficient, red cells display the morphologic stigmata of iron deficiency. This occurs despite the fact that intestinal iron absorption is markedly increased in response to a perceived need for iron for erythropoiesis. Nonhematopoietic tissues avidly assimilate the non-transferrin-bound metal. Similarly, mutant mice lacking tissue receptors for transferrin die during embryonic development from severe anemia, apparently resulting from ineffective iron delivery to erythroid precursor cells.
98Transferrin delivers its iron to developing normoblasts and other cells by binding to specific cell-surface receptors. The transferrin receptor (TFRC, TfR1) is a disulfide-linked homodimer of a glycoprotein with a single membrane-spanning segment and a short cytoplasmic segment. It is a type II membrane protein, with its N terminus located within the cell. The native molecular weight of TfR1 is ˜180 kDa. Each TfR1 homodimer can bind two transferrin molecules. Diferric transferrin is bound with higher affinity (association constant 2-7 × 10
-9 M) than monoferric transferrin. As a result, diferric transferrin has a competitive advantage in delivering iron to the erythroid precursors.
99 Apotransferrin has little affinity for the receptor at physiologic pH but considerable affinity at lower pH, an important factor in intracellular iron release.
Transferrin receptor 2 (TfR2), which shares ˜45% homology with TfR1 in its large extracellular domain,
100 binds transferrin with lower affinity, and its role in cellular iron uptake is unclear. TfR2 is highly expressed in the liver and, as discussed earlier in this chapter, acts as an iron sensor that regulates hepcidin expression.
36 TfR2 is also expressed in erythroid precursor cells, where it interacts with the erythropoietin receptor and enhances the delivery of EpoR to cell surface.
101The role of TfR1 is relatively well understood. TfR1 numbers are modulated during erythroid cell maturation, reaching their peak in intermediate normoblasts. Very few TfR1 molecules are found on burst-forming-unit erythroid cells, and only slightly greater numbers are found on colony-forming-unit erythroid cells. However, by the early normoblast stage, approximately 300,000 receptors are found on each cell, increasing to 800,000 at the intermediate stages. The rate of iron uptake is directly related to the number of receptors. The number decreases as reticulocytes mature, and late in maturation, erythroid cells shed all remaining receptors by exocytosis and by proteolytic cleavage.
102 The shed receptors (referred to as soluble transferrin receptors, sTfR) can be found in plasma in a concentration that correlates with the rate of erythropoiesis.
103 An increase in plasma sTfR is a sensitive indicator of erythroid mass and tissue iron deficiency.
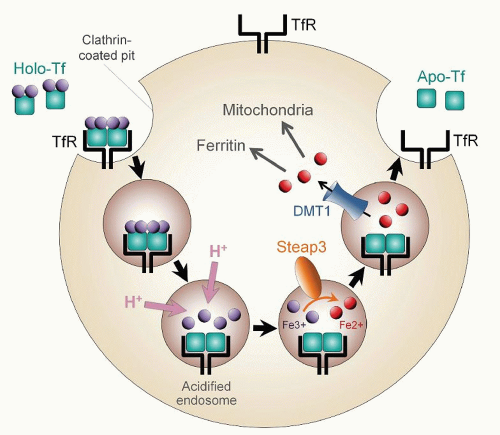
104,105After ligand and receptor interact, iron-loaded transferrin undergoes receptor-mediated endocytosis (
Fig. 23.5).
106 Specialized endocytic vesicles form, which are acidified to a pH of 5 to 6 by the influx of protons. The low pH facilitates release of iron from transferrin and strengthens the apotransferrin-receptor interaction. Released iron is reduced by an endosomal ferrireductase, STEAP3,
107 and transferred to the cytosol by DMT1.
108 Because DMT1 must cotransport protons with iron atoms, vesicle acidification is also important for the function of this transporter. After the iron enters the cytosol, the protein components of the endosome return to the membrane surface, where neutral pH promotes the release of apotransferrin to the plasma. Both transferrin and TfR1 participate in multiple rounds of iron delivery.
Mutations in DMT1 can have profound effect on erythroid development. Although rare, DMT1 mutations have been described in several patients with congenital iron deficiency anemia.
109,110,111 The patients suffer from severe microcytic, hypochromic anemia, due to the inability to transport iron out of Tf/TfR1 endocytic vesicles, thus preventing iron delivery for heme synthesis. Although DMT1 is also involved in duodenal iron absorption, most of these patients develop hepatic overload at a young age, suggesting that iron absorption is at least partially intact, probably due to dietary heme absorption. Blood transfusions to correct the anemia also contribute to patients’ iron overload.
Once inside the cell, erythroid iron must be shuttled to the mitochondrion, where it will be incorporated into protoporphyrin IX by ferrochelatase, the final enzyme of heme biosynthesis. How iron that is exported from endosome reaches the mitochondrion and is transported across the outer mitochondrial membrane is not well understood. Iron transport across the inner mitochondrial membrane seems to be mediated by mitoferrin-1, a transmembrane iron transporter.
112 Inactivation of the mitoferrin-1 gene in mice prevents red blood cell production, underscoring the importance of this protein in erythroid biology. An alternative mechanism for iron delivery from Tf/TfR endosome to the mitochondrion has also been proposed. The “kiss and run” hypothesis suggests that the Tf-containing endosome directly contacts the mitochondrion,
113 thereby bypassing the cytosol. The details of the mechanism remain to be determined.
The rate of iron entry into normoblasts is intimately related to heme biosynthesis.
114 In less mature erythrocyte precursors, an optimal amount of heme may be necessary to sustain TfR1 synthesis.
115 Furthermore, a decrease in intracellular free heme concentration induced by inhibitors of heme synthesis (e.g., isoniazid) leads to increased iron uptake, and an increase in intracellular free heme induced by inhibitors of globin synthesis (e.g., cycloheximide) has the opposite effect. These phenomena appear to reflect a feedback inhibition system that regulates the supply of iron according to the needs of the cell for hemoglobin synthesis. This is part of a highly coordinated erythroid regulatory system that acts to balance the amounts of
α– and
β-globin proteins and heme, avoiding toxic buildup of any intermediates of hemoglobin production.
116
Iron Metabolism within Normoblasts
In the normal subject, ˜80% to 90% of the iron that enters erythroid precursor cells is ultimately taken up by mitochondria and incorporated into heme. Most of the remainder is stored in ferritin.
117 Granules of ferritin may sometimes be detected using the Prussian blue reaction.
118 Normoblasts with Prussian blue-positive (siderotic) granules are called sideroblasts, and, if the granules persist after enucleation, the mature cells are called siderocytes. In normal individuals, approximately half of the normoblasts are sideroblasts, each containing less than five small granules. By electron microscopy, these normal siderotic granules are seen to be aggregations of ferritin, often surrounded by a membrane (siderosomes).
119 Isolated molecules of ferritin not surrounded by a membrane may also be seen by electron microscopy but not with light microscopy. Another kind of sideroblast, the ringed sideroblast, is found only under pathologic circumstances (sideroblastic anemias;
see Chapter 24). In the ringed sideroblast, siderotic granules form a full or partial ring around the nucleus, and electron microscopy reveals that the iron is deposited in mitochondria. The spleen plays an active role in the removal of the iron-laden mitochondria when cells enter the circulation.
It is intriguing that even though erythroid precursors require large amounts of iron and heme for hemoglobin synthesis, they have been found to express the heme exporter FLVCR and the iron exporter ferroportin. FLVCR, initially identified as the receptor for feline leukemia virus subgroup C which causes erythroid aplasia in cats, is highly expressed in early erythroid precursors. The loss of FLVCR in mice caused embryonic lethality due to the lack of definitive erythropoiesis.
120 When FLVCR was ablated postnatally, the mice developed a hyperchromic macrocytic anemia with proerythroblast maturation arrest. These studies suggest that FLVCR is necessary for survival of erythroid precursors. Because heme is toxic to cells at high concentrations, it is thought that FLVCR functions as a safety valve to prevent accumulation of excess heme early during erythroid differentiation.
Erythroid precursors also express ferroportin
86,121 but its role in erythroid maturation is yet unclear. It is interesting that erythroid cells preferentially express non-IRE ferroportin transcript during the stages of rapid iron uptake, thus avoiding any ironinduced increase in ferroportin translation dependent on the IRE/IRP system.
Macrophage Iron Recycling
Although there are many types of tissue macrophages, those that participate in the catabolism of red blood cells can be subdivided into two categories. One type, exemplified by pulmonary alveolar macrophages, is able to phagocytose erythrocytes or other cells and convert the iron they contain into storage forms, but lacks the ability to return the iron to the circulation. This type of macrophage appears to retain the iron throughout its life span. The second type of macrophage, comprising the reticuloendothelial system, acquires iron in a similar fashion but is able to return it to the plasma. The latter macrophages, found especially in the sinuses of the spleen and the liver, play a primary role in the normal reutilization of iron from destroyed red cells, allowing completion of the iron cycle shown in
Figure 23.4.
After erythrophagocytosis by reticuloendothelial macrophages, the red cell membrane is destroyed, heme is released from hemoglobin, and iron is liberated by heme oxygenase. The subcellular location of these events is not well defined. Subsequently, two phases of macrophage iron release are observed: an early phase, complete within a few hours after erythrophagocytosis, and a later phase, taking place over a period of days.
122 Iron is exported from macrophages through ferroportin, the iron exporter that is also found on the basolateral surface of absorptive enterocytes.
28 The early phase probably reflects immediate export of iron resulting from the sequential induction of ferroportin mRNA expression by heme released from hemoglobin, and ferroportin protein translation by heme-derived iron acting via the IRE/IRP system.
123 The later phase of iron release probably represents mobilization of stored iron in response to iron demand. This is likely achieved by modulating expression of hepcidin, the iron-regulatory hormone that causes degradation of macrophage ferroportin. Expression of hepcidin decreases when erythroid iron needs increase, allowing more ferroportin molecules to remain on the membrane of recycling macrophages.
124 Iron exported through ferroportin must be oxidized to be bound to transferrin. The ferroxidase reaction is catalyzed by the plasma copper protein, ceruloplasmin.
125 When the gene encoding ceruloplasmin was disrupted in mice, macrophages failed to release their iron at a normal rate, resulting in hypoferremia despite the presence of normal iron stores.
125 The abnormality was corrected by administering ceruloplasmin intravenously. A similar phenomenon was noted in aceruloplasminemic human subjects.
126 Ceruloplasmin probably acts by catalyzing the oxidation of ferrous iron to the ferric form, which is a prerequisite for iron binding by apotransferrin.
Because of the large daily iron flux through erythrophagocytosing macrophages (20 to 25 mg), iron release from macrophages is a critical determinant of the availability of iron for plasma transferrin and, consequently, for erythropoiesis. The anemia of inflammation, also called the anemia of chronic disease,
127 characterized by macrophage iron retention, illustrates the deleterious effects of perturbing the delicate balance between macrophage iron storage and release (
see Chapter 45).
Intracellular iron in macrophages is stored in two compounds: ferritin and hemosiderin. Iron-free ferritin (apoferritin) exists as a sphere with a diameter of 13 nm.
128 It has a hollow central cavity, 6 nm in diameter, which communicates with the surface by six channels through which iron and other small molecules can enter and leave. The protein shell is constructed of 24 molecules of two distinct ferritin subunits, designated H (for heavy or heart) and L (for light or liver), and these proteins are ubiquitously expressed. H and L ferritins are highly homologous, suggesting that they were derived from a common ancestral protein. However, there are functional differences between the two subunits. Loss of ferritin H in mice causes early embryonic lethality indicating that there is no functional redundancy between the two ferritin subunits.
129 H chains contain a ferroxidase center not found in L chains and are able to oxidize iron.
130,131 Ferritin shells rich in H chains reflect this property and acquire iron more rapidly,
132 whereas those rich in L chains appear to be more stable and resistant to denaturation. The primary role of ferritin is cellular protection against oxidative damage that would be caused by highly reactive free iron.
133At least 20 distinct ferritin heteropolymers, with varying proportions of L and H chains, have been isolated from human tissue.
134 Fully assembled, iron-free apoferritin has a molecular weight of approximately 400 to 500 kDa. When iron is
incorporated into the molecule, it is deposited in the central core. Theoretically, a single ferritin molecule could hold up to ˜4,500 iron atoms. More often, molecules with 2,000 or fewer iron atoms are found. The iron is stored as a trivalent polymer of ferric hydroxide and phosphate.
Although H and L chains make up the abundant cytoplasmic ferritin molecules, a unique ferritin protein is present in mitochondria, and is encoded by a distinct intronless gene.
135 Mitochondrial ferritin appears to have a ferroxidase activity, making it most analogous to H-ferritin.
136 Although incompletely understood, the role of mitochondrial ferritin is important enough that it has been conserved among species and can even be found in fruit flies.
137In a wide variety of cell types, the synthesis of apoferritin is stimulated by exposure to iron.
138 This effect of iron takes place at the translational level of protein synthesis, via the IRE/IRP system described earlier in the chapter. An iron-responsive element (IRE) found at the 5′ end of ferritin mRNAs is both necessary and sufficient for the iron effect.
139 In red cell precursors, hemin rather than iron itself may exert a similar function.
115,140Apoferritin is formed first, and iron is added later. A cytosolic iron chaperone, poly(rC)-binding protein 1 (PCBP1), delivers iron in the form of Fe
2+ to ferritin.
141 Iron then enters ferritin through one of the six penetrating channels. As it passes into the core, it is oxidized to the Fe
3+ form by molecular oxygen, with apoferritin catalyzing the oxidative process.
142 Iron is deposited irregularly and in varying amounts, forming microcrystals. Mobilization of iron from ferritin depends on iron reduction or chelation.
Ferritin is also found in plasma in small amounts (12 to 300
µg/L), and in the absence of inflammation,
143 the circulating ferritin concentration correlates roughly with the amount of iron in stores. Plasma ferritin appears to be actively secreted into plasma, primarily by macrophages
144,145 and is iron-poor compared to tissue ferritin.
Hemosiderin was formerly distinguished from ferritin on the basis of solubility and the Perls Prussian blue reaction. However, these distinctions are not reliable; as with hemosiderin, ferritin can also be stained with Prussian blue and can exist in insoluble forms. Hemosiderin is not a precisely definable chemical entity.
146 It appears to be formed by incomplete degradation of ferritin and conglomeration of iron, ferritin proteins, and other subcellular constituents. It differs from ferritin in having a higher iron-to-protein ratio as well as being less soluble in aqueous solutions. Standard hemosiderin preparations may contain a variety of organic constituents, including proteins, lipids, sialic acid, and porphyrins. In most instances, hemosiderin appears to be derived from ferritin. Hemosiderin appears to represent a more stable and less available form of storage iron than ferritin. Newly deposited or newly mobilized iron enters or leaves the ferritin compartment. Only after prolonged storage or continued mobilization does the hemosiderin compartment change in size.