Fig. 7.1
Classification of CLRs by intracellular signaling motifs. Representative CLRs are classified based on their intracellular signaling motifs: ITAM-containing receptors (such as Dectin-1, CLEC2, CLEC9A, and SIGN-R3), adaptor-recruiting receptors (such as Dectin-2, Mincle, MDL-1, and BDCA-2), ITIM-containing receptors (such as DCIR, CLEC12A, CLEC12B, and Ly49Q), and ITAM-ITIM-independent receptors (such as DC-SIGN, SIGN-R1, CLEC1, and LOX-1). See related review articles for more details (Geijtenbeek and Gringhuis 2009; Sancho and Reis e Sousa 2012). Abbreviations: SIGN-R3, DC-SIGN-related protein 3; MDL-1, myeloid DAP12-associating lectin 1; BDCA-2, blood dendritic cell antigen 2; DCIR, dendritic cell immunoreceptor; SIGN-R1, DC-SIGN-related protein 1; LOX-1, lectin-like oxidized low-density lipoprotein receptor 1
1.
Activation Syk-coupled CLRs: Spleen tyrosine kinase (Syk) is a tyrosine kinase involved in the signaling induced by a subset of CLRs. The coupling of CLRs to Syk can be direct via a single immunoreceptor tyrosine-based activation motif (ITAM) domain found in the cytoplasmic domain of some CLRs or indirect through the adaptors Fc receptor γ chain (FcRγ) or DNAX-activating protein of 12 kDa (DAP12), which bear classical Syk-recruiting ITAM motifs. Active Syk binds directly to several substrates, which in turn activate many downstream signaling pathways (Mocsai et al. 2010).
2.
Inhibitory CLRs with immunoreceptor tyrosine-based inhibition motif (ITIM) domains: Some CLRs express ITIM motifs that recruit phosphatases and thereby negatively regulate signaling through kinase-associated receptors, notably the Syk-coupled CLRs.
3.
CLRs without clear ITAM or ITIM domains: Although these CLRs can engage the endocytic machinery, the signaling pathways induced by these CLRs remain unclear.
Many pathogens possess atypical glycans that serve as ligands for CLRs, and some CLRs signal to potentiate microbicidal activity of myeloid cells and contribute to the activation of adaptive immunity (Geijtenbeek and Gringhuis 2009). The best example of this comes from the study of fungal infection. Fungal pathogens such as Candida albicans are recognized by multiple myeloid CLRs, including Dectin-1 (also known as C-type lectin domain family 7 member A, or CLEC7A), Dectin-2 (CLEC6A in human, CLEC4N in mouse), and Mincle (CLEC4E). Another example is Mycobacterium tuberculosis, which can be recognized by mannose receptor (MR) and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) (Mocsai et al. 2010; Tailleux et al. 2003; Kang et al. 2005). CLRs also detect molecules released from dead cells or exposed by cell corpses to induce or suppress inflammation. Such “self” ligands for CLRs include uric acids, F-actin, and a histone deacetylase complex subunit SAP130 (Shi et al. 2003; Neumann et al. 2014; Zhang et al. 2012; Ahrens et al. 2012; Yamasaki et al. 2008).
Although, as just described, the critical contribution of CLRs to detect pathogens has been extensively studied, it has been enigmatic as to whether and how CLRs are involved in tumor immunity. Very recently, it was found that Dectin-1 recognizes live tumor cells and triggers antitumor immune responses (Chiba et al. 2014). We therefore focus on Dectin-1 in the next section and discuss its roles in antitumor immunity.
7.1.3 Dectin-1
Dectin-1 is expressed in myeloid cells including dendritic cells and macrophages (Taylor et al. 2002) and works as a PRR for β-1,3-linked glucans present in the cell wall of fungi, bacteria, and plants (Brown 2006). Dectin-1 has an ITAM domain in the cytoplasmic tail and thus can directly activate Syk upon binding to agonistic ligands (Rogers et al. 2005; Underhill et al. 2005). Activated Syk, directly or indirectly, triggers the recruitment of the adaptor CARD9 to the membrane, resulting in activation of the IκB kinase (IKK) complex for canonical NF-κB signaling. Through activation of canonical NF-κB signaling, Dectin-1-Syk signaling induces secretion of cytokines, including IL-2, IL-6, IL-10, IL-23, and TNF-α (Sancho and Reis e Sousa 2012; Geijtenbeek and Gringhuis 2009). In addition, Dectin-1 can activate the noncanonical NF-κB pathway in CARD9-independent manner (Gringhuis et al. 2009). The Dectin-1-Syk axis also induces mitogen-activated protein kinase (MAPK) cascades and nuclear factor of activated T-cell (NFAT) signaling (LeibundGut-Landmann et al. 2007; Slack et al. 2007; Goodridge et al. 2007).
In addition to Syk activation, Dectin-1 induces a second signaling pathway mediated by the serine-threonine kinase Raf-1. Although Raf-1 does not depend on Syk signaling for its activation, Dectin-1-Raf-1 pathway converges with Syk-coupled pathways at the level of NF-κB activation. Dectin-1-induced phosphorylation of p65 at Ser276 subsequently leads to its acetylation. Acetylated p65 becomes transcriptionally active and then induces secretion of IL-6, IL-10, and IL-12 (Gringhuis et al. 2007, 2009).
7.2 The IRF Family of Transcription Factors as Regulators of PRR Signaling and Oncogenesis
The discovery of mammalian IRF family of transcription factors dates back to 1988, when a cDNA clone encoding a mouse protein that binds to a virus-inducible enhancer element of the IFNB gene was identified (Miyamoto et al. 1988). Since then, the IRF family has been shown to include nine members, IRF1–9, and these family members share significant homology within the conserved amino (N)-terminal DNA-binding domain (DBD) (Tamura et al. 2008; Yanai et al. 2012; Honda and Taniguchi 2006).
With the discovery of signal-transducing PRRs, IRFs have gained attention as essential regulators for the activation of immune cells (Honda and Taniguchi 2006; Kawai and Akira 2011; Geijtenbeek and Gringhuis 2009; Holm et al. 2013; Ikushima et al. 2013). It has been shown that certain microbial components, such as double-stranded RNA, LPS, and oligodeoxynucleotides containing unmethylated CpG motifs, activate the IRF-mediated induction of type I IFN genes through TLRs (Kawai and Akira 2011). In addition, the recent identification and characterization of receptors for cytosolic nucleic acids, such as cyclic GMP-AMP synthase (cGAS), stimulator of IFN genes (STING), IFN-γ-inducible gene 16 (IFI16), DNA-dependent activator of IRF (DAI), high-mobility group box proteins (HMGBs), retinoic acid-inducible protein I (RIG-I), and melanoma differentiation-associated gene 5 (MDA5), has revealed that these receptors also activate IRF3- and IRF7-dependent pathways for type I IFN induction (Ikushima et al. 2013; Sun et al. 2013; Burdette et al. 2011; Yin et al. 2012; Unterholzner et al. 2010; Takaoka et al. 2007; Yanai et al. 2009; Yoneyama et al. 2004; Kang et al. 2002; Holm et al. 2013).
Although IRFs were originally identified as transcriptional regulators of type I IFN and IFN-inducible genes, recent studies have revealed that IRFs are involved in other facets of PRR signaling. For example, IRF5 associates with MyD88 and regulates the induction of proinflammatory cytokine genes, such as TNF-α, IL-6, and IL-12p40 (Takaoka et al. 2005). Interestingly, recent findings have shown that this IRF5 function is inhibited by RIG-I-like receptor (RLR)-activated IRF3 (Negishi et al. 2012). IRF1, which is induced by IFN-γ, is another mediator that is activated by TLR-MyD88 pathway for the induction of specific genes such as IFN-β, inducible NO synthase (iNOS), and IL-12p35 (Kamijo et al. 1994; Liu et al. 2003; Ikushima et al. 2013). Further, IRF4 and IRF8 participate in TLR-mediated signaling in dendritic cells (Negishi et al. 2005; Tsujimura et al. 2004). Thus, many of the IRF members are essential regulators in PRR-mediated signaling. In addition, accumulating evidence indicates the oncogenic or anti-oncogenic function of IRFs in the regulation of cellular responses linked to oncogenesis (Table 7.1), for which detailed reviews are now available (Yanai et al. 2012). On the other hand, evidence is still limited about how IRFs, activated downstream of PRR signaling, contribute to the regulation of oncogenesis. Here, we focus below on IRF5, because of the recent finding for its involvement in Dectin-1 signaling in antitumor innate immune responses.
Table 7.1
A summary of oncogenic and anti-oncogenic roles of IRF
IRF | Function associated with oncogenesis | References | |
|---|---|---|---|
Oncogenic IRFs | IRF2 | Impairs p53 function | Pettersson et al. (2009) |
Promotes oncogenesis by antagonizing IRF1 (see below) | Harada et al. (1993) | ||
Nguyen et al. (1995) | |||
IRF4 | Promotes oncogenesis in multiple myeloma | Iida et al. (1997) | |
Heintel et al. (2008) | |||
Shaffer et al. (2008) | |||
Anti-oncogenic IRFs | IRF1 | Suppresses oncogene-induced transformation | Harada et al. (1993) |
Required for DNA damage-induced cell cycle arrest | Tanaka et al. (1996) | ||
Required for DNA damage-induced apoptosis | Pamment et al. (2002) | ||
Frontini et al. (2009) | |||
Tanaka et al. (1994) | |||
Tamura et al. (1995) | |||
Kano et al. (1999) | |||
IRF3 | Promotes DNA damage-induced apoptosis | Kim et al. (1999) | |
IRF5 | Suppresses oncogene-induced transformation | Yanai et al. (2007) | |
Required for DNA damage-induced apoptosis | Yanai et al. (2007) | ||
Hu et al. (2005) | |||
Required for Fas-induced apoptosis | Couzinet et al. (2008) | ||
Induces antitumor immune responses | Chiba et al. (2014) | ||
IRF6 | Promotes cell cycle arrest | Bailey et al. (2008) | |
IRF7 | Suppresses bone metastasis | Bidwell et al. (2012) | |
IRF8 | Inhibits myeloid cell growth | Hao and Ren (2000) | |
Tamura et al. (2003) | |||
Burchert et al. (2004) | |||
Dror et al. (2007) | |||
Promotes FAS-induced apoptosis | Hu et al. (2011) | ||
IRF9 | Stimulates p53 pathway | Takaoka et al. (2003) | |
Promotes DNA damage-induced responses | Weihua et al. (1997) |
Indeed, IRF5 has emerged as an interesting IRF family member in that it exerts tumor-suppressive function in two ways; first, it functions as an intrinsic tumor suppressor and mediator of antitumor innate immunity. It has been reported that IRF5 expression is suppressed in human leukemia and human ductal carcinoma and the attenuated expression of IRF5 correlates with disease stage (Barnes et al. 2003; Bi et al. 2011). In addition, a single point missense mutation (G202C), termed as IRF-5P68, was identified in peripheral blood cells from patients with adult T-cell leukemia/lymphoma (ATL) and chronic lymphocytic leukemia (CLL) (Yang et al. 2009). IRF-5P68 acts as a dominant negative regulator that interferes with IRF5 activity. These reports indicate that IRF5 inactivation relates to the development of human cancer. Ha-Ras-expressing Irf5 −/− MEFs show resistance to DNA damage-induced apoptosis and undergo transformation to form tumors in nude mice (Yanai et al. 2007). Interestingly, Irf5 mRNA is induced upon DNA damage by the tumor suppressor p53 (Yanai et al. 2007; Mori et al. 2002). However, since several p53 target genes, such as those encoding proapoptotic Puma and Noxa, are induced even in Irf5 −/− MEFs, IRF5 may act on an apoptotic pathway that is distinct from that of p53 (Yanai et al. 2007). Indeed, overexpression of IRF5 inhibits in vitro and in vivo B-cell lymphoma tumor growth in the absence of wild-type p53 (Barnes et al. 2003). Furthermore, ectopic expression of IRF5 sensitizes p53-proficient and p53-deficient colon cancer cells to DNA damage-induced apoptosis (Hu et al. 2005). Another antitumor function of IRF5 has been demonstrated; IRF5 activated by Dectin-1 signaling in dendritic cells and macrophages is critical to the enhancement of tumor-killing activity of natural killer (NK) cells, and this aspect is further elaborated below.
7.3 Antitumor Innate Immune Responses Through Dectin-1-IRF5 Axis
It has been well established that NK cells are essential effector cells of the innate arm of the immune system to control tumor progression by exerting their cytotoxicity (Yokoyama and Plougastel 2003). However, little has been known about whether or how other immune cells recognize tumor cells to assist NK cells for their tumoricidal activities.
During the course of the study on the role of IRF family members in antitumor immunity, it was found that B16F1 melanoma cells potently metastasize to lungs in Irf5 −/− , but not in Irf3 −/− or Irf7 −/− mice (Chiba et al. 2014). Bone marrow transplantation experiments revealed that IRF5 is required in cells of the hematopoietic origin for the control of tumor metastasis. It was found that dendritic cells and macrophages from splenocytes enhance tumor-killing activity of NK cells, wherein IRF5 in these cells, but not in NK cells, is required for the enhancement. Further, evidence was provided that these cells potentiate tumoricidal activity of NK cells via cell-to-cell contact, an observation consistent with the previous report showing the importance of direct cell contact between dendritic cells and NK cells in the enhancement of NK cell-mediated tumoricidal activity (Fernandez et al. 1999).
As such, these observations raise the interesting issue of whether and how the PRRs known to activate IRF5 are involved in oncogenesis. Of the pathways described for IRF5 activation, the TLR-MyD88 pathway is the best known (Takaoka et al. 2005; Ikushima et al. 2013). Nevertheless, the antitumor orchestration of dendritic cells, macrophages, and NK cells remains unaffected by MyD88 deficiency, suggesting that TLRs are not involved. Dectin-1 expressed by dendritic cells and macrophages activates IRF5 upon stimulation by curdlan, a fungi-derived β-1,3 polysaccharide, which leads to the induction of type I IFNs for antifungal immunity (del Fresno et al. 2013). When Dectin-1-mediated IRF5 activation was examined in splenocytes by B16F1 melanoma cells, nuclear translocation of IRF5, a hallmark of its activation (Takaoka et al. 2005; Ikushima et al. 2013), was observed in wild-type splenocytes but not in splenocytes from Dectin-1-deficient mice (Clec7a −/− mice). Thus, live tumor cells can activate Dectin-1-dependent IRF5 activation (Chiba et al. 2014). Furthermore, enhancement of NK cell-mediated tumoricidal activity by dendritic cells and macrophages was attenuated when splenocytes from Clec7a −/− mice were used in lieu of those from wild-type mice. Expectedly, a marked enhancement of metastasis of B16F1 cells was seen in the lungs of Clec7a −/− mice, which was even more pronounced as compared to Irf5 −/− mice; the enhanced tumor growth was also observed when mutant mice were inoculated subcutaneously with B16F1 cells.
These results revealed that Dectin-1 in innate immune cells directly recognizes tumor cells to activate IRF5 and other signaling pathways, which mediates antitumor activity by NK cells. Indeed, in addition to B16F1 cells, Dectin-1 binds strongly to several mouse and human cancer cell lines but weakly to non-transformed cells (Chiba et al. 2014) (Fig. 7.2). Since the Dectin-1 binding to B16F1 cells is markedly reduced upon N-glycosidase treatment, there is a major requirement for N-glycan structures to Dectin-1 binding. Consistently, the tumoricidal activity of splenocytes was markedly reduced for the N-glycosidase-treated B16F1 cells (Chiba et al. 2014). These data strongly indicate that Dectin-1 recognizes N-glycan structures overexpressed in those tumor cells, leading to the activation of IRF5 and other pathways. However, the precise recognition structure needs to be clarified further. The critical target genes of the Dectin-1 pathway in dendritic cells and macrophages, which contribute to the enhancement of NK activity by these cells, also need to be identified.
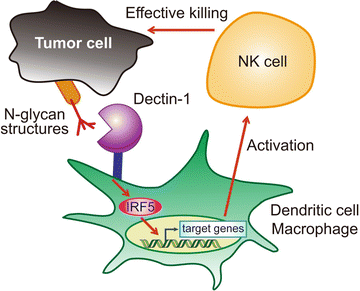
Fig. 7.2
Activation of NK cell-mediated tumor killing through recognition of tumor cells by Dectin-1. Dectin-1 expressed on dendritic cells and macrophages critically contributes to the enhancement of NK cell-mediated killing of tumor cells. IRF5 is activated by Dectin-1 signaling in these immune cells, and this Dectin-1-IRF5 pathway is required for effective tumoricidal activity of NK cells. Tumor cell-mediated Dectin-1 signaling is instigated by receptor recognition of N-glycan structures on the surface of some but not all tumor cells, which we propose to term tumor-associated molecular patterns (TAMPs)
7.4 Implications and Future Perspectives
Although the recognition of tumor-specific antigens by lymphocytes of the adaptive immune system has been studied extensively (Restifo et al. 2012), data for the role of innate PRRs in antitumor innate responses was scarce. The critical role of Dectin-1 (and its downstream activation of IRF5) in the orchestration of antitumor innate immune responses is the first demonstration that an innate immune receptor contributes to antitumor recognition and signaling, offering new insights into the NK cell-mediated antitumor activity of the innate immune system. Although yet to be fully investigated, it is possible that recognition of N-glycan structures by Dectin-1 is contingent on the absolute expression levels of N-glycans and/or their associated proteins on the cell; that is, tumor cells with “increased self” molecules are targets for innate immune recognition for the activation of the immune system. On the other hand, it is also plausible that recognition by Dectin-1 requires particular tumor-specific N-glycan-containing structures, which may belong to “altered self” (Medzhitov and Janeway 2002). Whichever the case, Dectin-1-binding structures may fit into the category of “tumor-associated molecular patterns (TAMPs)” vis-à-vis PAMPs for invading pathogens and DAMPs for normal cells subjected to stress or death (Rubartelli and Lotze 2007). This issue obviously merits more advanced study. In this regard, an interesting future issue is whether other members of the CLR family may also contribute to the antitumor innate immune system by the recognition of other TAMPs.
Since tumor cells are phenotypically and functionally heterogeneous within the tumor mass (Meacham and Morrison 2013), tumor cells may show a differential expression profile for Dectin-1 ligands in vivo. If this is the case, one may envisage that the in vivo progression of a tumor is controlled via direct and indirect Dectin-1 signaling in that NK cells activated by dendritic cells and macrophages via the receptor signaling may exert tumoricidal activities on tumor cells regardless of their Dectin-1 ligand expression. In light of the well-accepted tenet that innate immunity instructs adaptive immunity (Janeway and Medzhitov 2002), an interesting future issue is whether Dectin-1 signaling by tumor recognition also affects antitumor adaptive immune responses and also may provide new means for the efficient immune responses for cancers such as Dectin-1 agonistic antibodies. Thus, the study on the innate receptor signaling and downstream signaling mechanisms in the regulation of tumor generation and development may provide a new avenue of tumor immunology, particularly in combination with the harnessing antitumor adaptive immunity.
Acknowledgments
We would like to apologize to all of the authors whose invaluable work we could not discuss or cite in this chapter due to space constraints. Our work was supported in part by the Core Research for Evolutional Science and Technology (CREST) of JST, a Grant-in-Aid for Exploratory Research of MEXT, and a Grant-In-Aid for Scientific Research on Innovative Areas of MEXT.
References
Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, Batista F, Thompson B, Way M, Reis e Sousa C, Schulz O (2012) F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36(4):635–645. doi:10.1016/j.immuni.2012.03.008 CrossRefPubMed
Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207(5):1045–1056. doi:10.1084/jem.20100050 CrossRefPubMedCentralPubMed
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13(9):1050–1059. doi:10.1038/nm1622 CrossRefPubMed
Bailey CM, Abbott DE, Margaryan NV, Khalkhali-Ellis Z, Hendrix MJ (2008) Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Mol Cell Biol 28(7):2235–2243. doi:10.1128/MCB. 01866-07 CrossRefPubMedCentralPubMed
Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM (2003) Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res 63(19):6424–6431PubMed
Bi X, Hameed M, Mirani N, Pimenta EM, Anari J, Barnes BJ (2011) Loss of interferon regulatory factor 5 (IRF5) expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Breast Cancer Res 13(6):R111. doi:10.1186/bcr3053 CrossRefPubMedCentralPubMed
Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, de Weerd NA, Gould J, Argani P, Moller A, Smyth MJ, Anderson RL, Hertzog PJ, Parker BS (2012) Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 18(8):1224–1231. doi:10.1038/nm.2830 CrossRefPubMed
Blasius AL, Beutler B (2010) Intracellular toll-like receptors. Immunity 32(3):305–315. doi:10.1016/j.immuni.2010.03.012 CrossRefPubMed
Brown GD (2006) Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 6(1):33–43. doi:10.1038/nri1745 CrossRefPubMed
Related posts:
 In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
 Hepatitis C Virus (HCV)-Induced Inflammation: The Role of Cross-Talk Between HCV-Infected Hepatocytes and Stellate Cells
Hepatitis C Virus (HCV)-Induced Inflammation: The Role of Cross-Talk Between HCV-Infected Hepatocytes and Stellate Cells
 Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
 Role of Bone Marrow-Derived CCR1+ Cells in Colon Cancer Invasion and Metastasis
Role of Bone Marrow-Derived CCR1+ Cells in Colon Cancer Invasion and Metastasis
 Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
 Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


