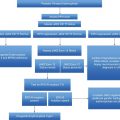
Diagnostic algorithm for hypereosinophilia and treatment options based on each subtype. FISH, fluorescent in situ hybridization; RT-PCR, reverse transcriptase polymerase chain reaction; NOS, not otherwise specified; WHO, World Health Organization; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CEL, chronic eosinophilic leukemia.
Testing for secondary causes of eosinophilia typically involves stool ova and parasite testing, and sometimes stool culture/antibody testing for specific parasites (e.g., Strongyloides). The type and frequency of laboratory and imaging tests (e.g., chest X-ray, electrocardiogram and echocardiography, computed tomography (CT) scan of the chest, abdomen/pelvis) are guided by the patient’s travel history, symptoms, and findings on physical examination. For patients with eosinophilia and signs/symptoms related to lung disease, pulmonary function testing, bronchoscopy with lavage or biopsy, and serologic tests (e.g., Aspergillus IgE to evaluate for allergic bronchopulmonary aspergillosis) may also be considered.
The internist and a consultant in infectious diseases identify no cause of the eosinophilia. The patient is referred to hematology, but misses the appointment. He returns 9 months later with complaints of worsening fatigue, dyspnea on exertion, and weight gain. On physical examination, an S3 murmur is auscultated, the spleen is palpated 5 cm below the left costal margin, and lower-extremity edema is present. The current CBC reveals a WBC count of 37 × 109/L with 38% eosinophils (AEC ~14.6 × 109/L). Myeloid immaturity is not present. An echocardiogram reveals a decreased ejection fraction of 35%. Endomyocardial biopsy reveals an extensive eosinophilic infiltrate. No new reactive causes of hypereosinophilia have emerged.
Question 2. Does the patient currently meet criteria for idiopathic hypereosinophilic syndrome (HES)?
A. Yes
B. No.
Answer: B.
Idiopathic HES is a diagnosis of exclusion defined by the following criteria: (1) an AEC >1.5 × 109/L lasting for more than 6 months; (2) signs of organ damage; and (3) other causes of eosinophilia have been ruled out.12 Although no obvious causes of reactive eosinophilia have emerged, a workup for primary (clonal) eosinophilia has not yet been undertaken.
The 2008 World Health Organization (WHO) classification of myeloid neoplasms helps to frame the approach for evaluating primary eosinophilias. In this WHO scheme, a new major category was added, “Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of platelet-derived growth factor receptor alpha (PDGFRA), platelet-derived growth factor receptor beta (PDGFRB), or fibroblast growth factor receptor 1 (FGFR1)” (Table 13.1).13 “Chronic eosinophilic leukemia – not otherwise specified” (CEL-NOS) is another primary eosinophilic neoplasm subsumed within the WHO category of myeloproliferative neoplasms (MPNs) (Table 13.1).14 CEL-NOS is defined by absence of the Philadelphia chromosome or a rearrangement involving PDGFRA/B and FGFR1. It also excludes other WHO-defined acute and chronic myeloid neoplasms that may be associated with eosinophilia (see below). CEL-NOS is characterized by an increase in blasts in the bone marrow or blood (but fewer than 20% to exclude acute leukemia as a diagnosis), and/or there is evidence for a non-specific cytogenetic abnormality (e.g., trisomy 8) or other clonal marker. To date, no recurrent genetic abnormalities have been identified in CEL-NOS, with the exception of a recent report indicating some cases with recurrent KIT M541L mutations.15 If none of the aforementioned eosinophilic diseases is uncovered (including lymphocyte-variant hypereosinophilia: see below), the diagnosis of idiopathic hypereosinophilia (organ damage absent) or idiopathic HES (organ damage present) can be rendered.
1. Acute myeloid leukemia and related disorders 2. Myeloproliferative neoplasms (MPN)
3. Myelodysplastic syndromes (MDS)
4. MDS/MPN
5. Myeloid and lymphoid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1
|
The requirement that hypereosinophilia endure for at least 6 months is no longer felt to be a necessary criterion for HES. The rapid pace and more sophisticated tools now available to evaluate hypereosinophilia, or the need for immediate treatment, may negate this requirement. In some patients, HES may begin as a provisional diagnosis because a definitive etiology for hypereosinophilia emerges.4
Question 3. Which of the following genetic markers should be obtained from the peripheral blood as part of the initial workup of primary eosinophilia in this patient?
A. JAK2 V617F
B. BCR-ABL
C. Fluorescent in situ hybridization (FISH) for the CHIC2 deletion
D. Calreticulin (CALR) exon 9 mutation
Answer: C, Fluorescent in situ hybridization (FISH) for the CHIC2 deletion.
Clues to the presence of a primary eosinophilia may emerge from the evaluation of the blood smear. Review of the peripheral smear for circulating blasts, dysplastic cellular morphology, monocytosis, and elevated serum B12 or serum tryptase level(s) in conjunction with bone marrow morphologic, cytogenetic, and immunophenoytpic analysis will help identify whether a WHO-defined eosinophilia-associated myeloid neoplasm is present. Examples include acute myelogenous leukemia (AML) (especially inv(16)(p13q22) or t(16;16)(p13;q22)), myelodysplastic syndrome (MDS), systemic mastocytosis, MPNs (chronic myeloid leukemia, polycythemia vera, essential thrombocythemia, and primary myelofibrosis), and MDS/MPN overlap disorders (e.g. chronic myelomonocytic leukemia) (Table 13.2).14
| Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1 |
| Diagnostic criteria of a myeloproliferative neoplasm (MPN)a with eosinophilia associated with FIP1L1-PDGFRA |
| A myeloproliferative neoplasm with prominent eosinophilia and |
| Presence of a FIP1L1-PDGFRA fusion geneb |
| Diagnostic criteria of MPN associated with ETV6-PDGFRB fusion gene or other rearrangement of PDGFRB |
| A myeloproliferative neoplasm, often with prominent eosinophilia and sometimes with neutrophilia or monocytosis and |
| Presence of t(5;12)(q31~q33;p12) or a variant translocationc or, demonstration of an ETV6-PDGFRB fusion gene or rearrangement of PDGFRB |
| Diagnostic criteria of MPN or acute leukemia associated with FGFR1 rearrangement |
| A myeloproliferative neoplasm with prominent eosinophilia and sometimes with neutrophilia or monocytosis |
| Or |
| Acute myeloid leukemia or precursor T-cell or precursor B-cell lymphoblastic leukemia/lymphoma (usuallyassociated with peripheral blood or bone marrow eosinophilia) |
| And |
| Presence of t(8;13)(p11;q12) or a variant translocation leading to FGFR1 rearrangement demonstrated in myeloid cells, lymphoblasts, or both |
| Chronic eosinophilic leukemia, not otherwise specified (NOS) |
| 1. There is eosinophilia (eosinophil count >1.5 × 109/L) |
| 2. There is no Philadelphia chromosome or BCR-ABL fusion gene or other myeloproliferative neoplasms (polycythemia vera, essential thrombocythemia, primary myelofibrosis, systemic mastocytosis) or myelodysplastic syndrome (MDS)/MPN (chronic myelomonocytic leukemia or atypical chronic myeloid leukemia) |
| 3. There is no t(5;12)(q31~q35;p13) or other rearrangement of PDGFRB |
| 4. There is no FIP1L1-PDGFRA fusion gene or other rearrangement of PDGFRA |
| 5. There is no rearrangement of FGFR1 |
| 6. The blast cell count in the peripheral blood and bone marrow is less than 20% and there is no inv(16)(p13q22) or t(16;16)(p13;q22) or other feature diagnostic of acute myeloid leukemia (AML) |
| 7. There is a clonal cytogenetic or molecular genetic abnormality, or blast cells are more than 2% in the peripheral blood or more than 5% in the bone marrow |
a Patients presenting with acute myeloid leukemia or lymphoblastic leukemia/lymphoma with eosinophilia and an FIP1L1-PDGFRA fusion gene are also assigned to this category.
b If appropriate molecular analysis is not available, this diagnosis should be suspected if there is a Philadelphia-negative MPN with the hematological features of chronic eosinophilic leukemia associated with splenomegaly, a marked elevation of serum vitamin B12, elevation of serum tryptase, and increased bone marrow mast cells.
c Because t(5;12)(q31~q33;p12) does not always lead to an ETV6-PDGFRB fusion gene, molecular confirmation is highly desirable. If molecular analysis is not available, this diagnosis should be suspected if there is a Phildalephia-negative MPN associated with eosinophilia and with a translocation with a 5q31-33 breakpoint.
Laboratory evaluation of primary eosinophilia should begin with screening of the peripheral blood for the FIP1L1-PDGFRA gene fusion by reverse transcriptase polymerase chain reaction (RT-PCR) or interphase/metaphase FISH.16,17 Many clinical laboratories now employ probes that hybridize to the region between the FIP1L1 and PDGFRA genes where the CHIC2 gene is located; deletion of CHIC2 is a surrogate for the cytogenetically occult 800-kb deletion on chromosome 4q12 that results in the fusion of FIP1L1 and PDGFRA.17 If testing for FIP1L1-PDGFRA is not available, evaluation of the serum tryptase level may be a useful surrogate test since increased levels have been associated with the presence of the FIP1L1–PDGFRA fusion or other MPNs associated with hypereosinophilia.18 Genetic rearrangement of PDGFRA (fusion partners besides FIP1L1), PDGFRB, and FGFR1 can usually be inferred by their associated abnormal karyotype rearrangement of 4q12 (PDGFRA), 5q31-33 (PDGFRB) or 8p11-13 (FGFR1).13 Over 20 gene fusion partners of PDGFRB have been described.8 Eosinophilic myeloid neoplasms related to fusions involving the FGFR1 gene are similarly rare.13 In these cases, the association of t(8p11-13) breakpoint with lymphoblastic lymphoma with eosinophilia and myeloid hyperplasia was first described in 1995,19 and since 1998, more than 10 fusion partners of FGFR1 have been reported,8 with ZNF198-FGFR1 being the most common.
Although eosinophilia can accompany BCR-ABL-positive chronic myeloid leukemia (and acute lymphoblastic leukemia), as well as JAK2 V617F-positive MPNs, there are no other clinicopathologic findings in this particular case to steer the physician to these diagnoses. FLT3 mutations (ITD or D835) are found primary in AML and confer a worse prognosis. Interestingly, myeloid or lymphoid neoplasms with eosinophilia and reciprocal translocations involving FLT3 (e.g., ETV6-FLT3) or JAK2 (e.g., PCM1-JAK2) have been published20,21 and in such cases the use of FLT3 and JAK inhibitors, respectively, is now being investigated.
Treatment of subtypes of hypereosinophilia
Case 1: part 2
The patient undergoes peripheral blood testing with FISH for the CHIC2 deletion that is positive in 84/200 cells (42%).
Question 4. What treatment should be initiated?
A. Hydroxyurea
B. Corticosteroids (prednisone 1 mg/kg)
C. Imatinib
D. Imatinib + prednisone 1 mg/kg
Answer: D, Imatinib + prednisone 1 mg/kg.
Imatinib is considered the standard of care in patients with FIP1L1-PDGFRA-positive disease, and the rare patients with alternate PDGFRA fusions or rearranged PDGFRB. The hematologic benefit of imatinib in myeloid neoplasms associated with eosinophilia was characterized in early studies22–24 before the therapeutic target FIP1L1-PDGFRA was identified by Cools et al.16 Molecular remissions by PCR testing of the peripheral blood were reported by the National Institutes of Health in 5 of 6 FIP1L1-PDGFRA-positive patients after 1–12 months of imatinib.25 Numerous reports have now described rapid induction of molecular remissions in imatinib-treated FIP1L1-PDGFRA-positive patients. Although 100 mg daily may be sufficient to achieve a molecular remission in most patients, others may require higher maintenance doses in the range of 300–400 mg daily. Dosing of 100–200 mg weekly may be sufficient to maintain a molecular remission in some patients.26 However, it is not clear what is the optimal maintenance dose of imatinib to sustain long-term molecular remissions.
Longer-term outcomes of imatinib-treated patients with FIP1L1-PDGFRA-positive eosinophilic myeloid neoplasms were evaluated in an Italian prospective cohort of 27 patients with a median follow-up period of 25 months (range 15–60 months).27 Patients were dose-escalated from an initial dose of 100 mg daily to a final dose of 400 mg daily. Complete hematologic remission was achieved in all patients within 1 month, and all patients became PCR-negative for FIP1L1-PDGFRA after a median of 3 months of treatment (range 1–10 months). Patients continuing imatinib remained PCR-negative during a median follow-up period of 19 months (range 6–56+ months). Another European study prospectively assessed the natural history of molecular responses to imatinib doses of 100–400 mg daily.28 Among 11 patients with high pretreatment transcript levels, all achieved a 3-log reduction in transcript levels by 1 year of therapy, and 9 of 11 patients achieved a molecular remission.
In patients with rearrangements of PDGFRB or PDGFRA variants other than FIP1L1-PDGFRA, imatinib, usually at doses of 400 mg daily, can elicit durable hematologic and cytogenetic remissions,29 reviewed in30. Similar to FIP1L1-PDGFRA, FISH can be used to assess response to imatinib in PDGFRB-rearranged cases.
Cardiogenic shock has been reported in a few FIP1L1-PDGFRA-positive patients after initiation of imatinib.31,32 It is therefore recommended that corticosteroids be utilized during the first 7–10 days of imatinib therapy in patients with known cardiac disease and/or elevated serum troponin levels which may indicate eosinophil-mediated heart damage or other cardiac comorbidities.32 In this patient with a biopsy-proven cardiac eosinophilic infiltrate and signs of heart failure, it would be prudent to add prednisone with initiation of imatinib; prednisone can be quickly tapered if there is no evidence of cardiac decompensation.
In patients like this, with established tissue damage, the frequency of serial evaluations is determined by the severity and extent of organ compromise, and/or by worsening of the AEC or organ function. However, in the absence of organ disease, no consensus guidelines exist to guide when to initiate treatment based on a specific AEC. Algorithms have incorporated serial monitoring of eosinophil counts, bone marrow aspiration and biopsy with cytogenetics, evaluation of T-cell clonality/immunophenotyping, and directed organ assessment (e.g., chest X-ray and/or CT thorax, echocardiography, cardiac troponin, pulmonary function testing, endoscopy, skin biopsy) in order to identify occult organ disease.
Case 1: part 3
The patient commences imatinib 100 mg daily and achieves a complete hematologic remission within 1 month. Splenomegaly has resolved, and he is maintained on medical therapy for heart failure with a repeat echocardiogram showing an ejection function of 45%. After 3 months, FISH testing for the CHIC2 deletion from the peripheral blood is negative. The patient is lost to follow-up. He returns to clinic 1 year later with complaints of night sweats, a 20 pound weight loss, and progressive dyspnea on exertion. The patient indicates he stopped imatinib 6 months earlier because he felt well. On exam, palpable splenomegaly 15 cm below the left costal margin is noted. A repeat echocardiogram shows a restrictive cardiomyopathy with an ejection function of 28%. The CBC shows loss of hematologic remission with a WBC count of 54 × 109/L, hemoglobin 8.5 g/dL, and platelet count 94 × 109/L. The differential reveals 15% neutrophils, 5% bands, 10% lymphocytes, 52% eosinophils, 12% immature myeloids (myelocytes, metamyelocytes), and 6% blasts. The smear reveals occasional teardrop and nucleated red blood cells. A bone marrow biopsy shows marked hypercellularity and eosinophilia, 8% myeloblasts, and MF-2 reticulin fibrosis. Cytogenetics reveal trisomy 8 and del (20q).
A. Switch to prednisone
B. Multiagent AML-type (idarubicin/cytarabine) chemotherapy
C. JAK2 V617F mutation testing
Answer: D, Sequence analysis of PDGFRA to evaluate for resistance mutation(s).
Despite in-depth and durable molecular remissions, discontinuation or non-compliance with imatinib may lead to disease relapse. In a dose de-escalation trial of imatinib in 5 patients who had achieved a stable hematologic and molecular remission at 300–400 mg daily for at least 1 year, molecular relapse was observed in all patients after 2–5 months of either imatinib dose reduction or discontinuation.33 Molecular remissions were re-established with reinitiation of imatinib in all patients at a dose range of 100–400 mg daily. In a cohort of patients evaluated by the Mayo Clinic, hematologic relapse occurred only several weeks after discontinuation of imatinib in 4 patients.34 These data indicate that imatinib does not cure FIP1L1-PDGFRA–positive neoplasms and argue for indefinite imatinib treatment.
FIP1L1-PDGFRA-positive patients can develop resistance to imatinib; most cases involve the T674I mutation within the ATP-binding domain of PDGFRA.16,35,36 T674I PDGFRA is analogous to the T315I ABL1 mutation in chronic myeloid leukemia, which confers pan-resistance to the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. However, unlike chronic myeloid leukemia, secondary resistance is very rare, and is usually observed during advanced phases of the disease. The presence of additional cytogenetic abnormalities, such as in this case, may confer resistance; however, this should not dissuade one from looking for resistance mutations within the PDGFRA gene.
Options for second-line treatment for T674I imatinib resistance are limited. One patient with the FIP1L1-PDGFRA T674I mutation in blast crisis responded briefly to sorafenib, but this was followed by rapid emergence of a pan-resistant FIP1L1-PDGFRA D842V mutant.37 Additional reports have demonstrated either in vitro or in vivo activity of sorafenib, midostaurin (PKC412), or nilotinib against the T674I mutant.38–41 The ability of alternative tyrosine kinase inhibitors to elicit durable clinical remissions (despite in vitro data demonstrating inhibitory activity against mutated fusions) has been disappointing.42
In this case, it would be premature to use induction chemotherapy since the patient does not have a diagnosis of AML; however, the patient is demonstrating signs of disease progression that may soon require higher-intensity therapy, such as induction chemotherapy or allogeneic transplantation if a suitable donor can be identified. However, the patient’s cardiac function may preclude such options. Although the patient has marked splenomegaly and marrow fibrosis, his diagnosis is not myelofibrosis, and the JAK2 V617F mutation is unlikely to be present. Steroids will not be helpful in this clonal myeloid disorder showing evolution towards AML.
Related posts:
 Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
 Incorporating symptomatic assessment in therapy choice for patients with myeloproliferative neoplasms
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
Incorporating symptomatic assessment in therapy choice for patients with myeloproliferative neoplasms
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
 Women and children with myeloproliferative neoplasms – special considerations
Women and children with myeloproliferative neoplasms – special considerations
 Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


