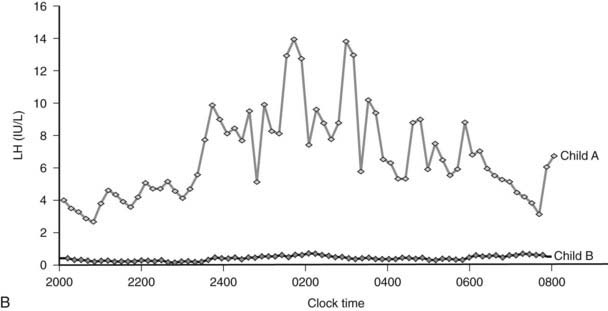
FIGURE 29-1. A, Clinical response to leptin therapy in congenital leptin deficiency. B, Leptin therapy is associated with pulsatile gonadotropin secretion at an appropriate developmental age in child A (age 11 years) compared with child B (age 5 years).
LEPTIN RECEPTOR DEFICIENCY
Up to 3% of patients with severe obesity have been found to harbor mutations in the leptin receptor gene that are associated with loss of function in vitro as measured by an inability to phosphorylate STAT3.66 Although heterozygosity for LEP or LEPR mutations is associated with an increase in body weight,66,67 severe obesity requires the loss of two alleles as the result of homozygous or compound heterozygous mutations. Serum leptin levels are not elevated disproportionately in LEPR deficiency,66 although particular mutations located near the transmembrane domain can result in a truncated extracellular domain that may act as a false binding protein, resulting in abnormally elevated leptin levels.68,69 The clinical phenotype of congenital leptin receptor deficiency is similar to that of leptin deficiency, with hyperphagia, severe early-onset obesity, hypogonadism, and frequent infections.66
POMC DEFICIENCY
Leptin activates hypothalamic neurons expressing pro-opiomelanocortin (POMC), and this action is functionally important for leptin’s action on appetite and body weight.70 In 1998, Krude and colleagues reported two unrelated obese German children who were homozygous or compound heterozygous for mutations in POMC (pro-opiomelanocortin)71; subsequently, several other children have been reported.72,73 Initial presentation occurs in neonatal life with adrenal crisis due to adrenocorticotropic hormone (ACTH) deficiency (POMC is a precursor of ACTH in the pituitary), and children require lifelong glucocorticoid replacement. Children have pale skin and red hair caused by the lack of melanocyte-stimulating hormone (MSH) action at melanocortin 1 receptors in the skin and hair follicles,72 although hypopigmentation may be less obvious in children from different ethnic backgrounds.73 POMC deficiency leads to hyperphagia and early-onset obesity, caused by loss of melanocortin signaling at the melanocortin 4 receptor (MC4R). Although, as yet, no specific treatment is available, selective small molecule MC4R agonists are being developed, and it is likely that these children would be highly responsive to such agents. It is notable that a high prevalence of overweight/obesity has been observed in the heterozygous relatives of children with complete POMC deficiency,72,73 suggesting that heterozygous point mutations in POMC might contribute to inherited obesity.74–76
PROHORMONE CONVERTASE 1 DEFICIENCY
Three unrelated subjects with compound heterozygous or homozygous mutations in the prohormone convertase (PC1/3) gene have been described.77–79 PC1/3 encodes an enzyme involved in the posttranslational processing of multiple prohormones. A prominent clinical feature is severe small intestinal absorptive dysfunction in neonatal life, which is likely to be attributable to impaired prohormone processing within the enteroendocrine cells and nerves that express PC1 throughout the gut.78 Postprandial hypoglycemia due to impaired processing of proinsulin to mature insulin is a key clinical feature and serves as the basis for a convenient diagnostic test, as elevated plasma levels of proinsulin and 32/33 split proinsulin in the context of low levels of mature insulin are found in PC1-deficient patients.78 Hypogonadotropic hypogonadism due to impaired processing of preprogonadotropin-releasing hormone contributes to infertility, and impaired processing of POMC contributes to hyperphagic severe early-onset obesity.
MELANOCORTIN 4 RECEPTOR DEFICIENCY
Mutations in MC4R have been reported in up to ≈6% of patients with severe early-onset obesity80 and are found at a frequency of approximately 1 in 1000 in the general UK population,81 making this one of the most common human monogenic diseases. Although we found a 100% penetrance of early-onset obesity in heterozygous probands, others have described obligate carriers who were not obese.82 Given the large number of potential influences on body weight, perhaps it is not surprising that both genetic and environmental modifiers will have important effects in some pedigrees. When all of these observations are considered, codominance, with modulation of expressivity and penetrance of the phenotype, is the most appropriate descriptor for the mode of inheritance.83
Affected subjects exhibit hyperphagia; this is not as severe as that seen in leptin deficiency, although it often starts in the first year of life. Of particular note is the finding that the severity of receptor dysfunction seen in in vitro assays can predict the amount of food ingested at a test meal by the subject who is harboring that particular mutation.80 An elevated respiratory quotient (ratio of carbohydrate to fat oxidation) in MC4R deficiency is consistent with an impaired ability to mobilize fat, as has been seen in MC4R knockout mice.84 Alongside the increase in fat mass, MC4R-deficient subjects also have an increase in lean mass that is not seen in leptin deficiency and a marked increase in bone mineral density. Linear growth of these subjects is striking, with affected children having a height standard deviation score (SDS) of +2 compared with population standards, and adults having an increased final height when compared with equally obese adults with a normal MC4R genotype. MC4R-deficient subjects also have higher levels of fasting insulin than age- and BMI SDS–matched children.
Diagnosis and Treatment
MC4R is a single exon gene that is readily amenable to rapid nucleotide sequencing, thus genetic analysis could be performed as a routine diagnostic test. However, before any causality is attributed to a mutation, it is necessary in our view to (1) establish that the sequence variant is absent from a panel of ethnicity-matched control subjects of appropriate number (≥100 alleles), (2) confirm that it results in impaired signaling when studied in vitro, and (3) determine that it co-segregates with obesity in family members. Although, at present, no specific therapy is known for MC4R deficiency, it is highly likely that these subjects would respond well to pharmacotherapy that overcomes the reduction in hypothalamic-melanocortinergic tone that is seen in these patients. Because most patients are heterozygotes with one functional allele intact, it is possible that small molecule MC4R agonists might, in the future, be excellent candidate drugs for this disorder.
OTHER SYNDROMES
Holder and colleagues studied a girl with hyperphagia and early-onset obesity and a balanced translocation between 1p22.1 and 6q16.2.85 The translocation disrupted the SIM1 gene on 6q. The Drosophila single-minded (Sim) gene is a regulator of neurogenesis, and in the mouse, Sim1 is expressed in the developing central nervous system and is essential for formation of the supraoptic and paraventricular (PVN) nuclei, which express the melanocortin 4 receptor. Mice heterozygous for loss of function mutations in Sim1 are obese, making it very likely that disruption of this gene in the child was the cause of her obesity.86 A number of patients with obesity, hypotonia, and developmental delay in association with interstitial chromosome 6q deletions have been described, although whether this syndrome can be attributed to SIM1 remains unclear.87
The WAGR syndrome (Wilms tumor, anorexia, ambiguous genitalia, and mental retardation) is one of the best studied contiguous gene syndromes associated with chromosomal deletions at 11p13, the location of the WT1 gene.88 A few patients with WAGR syndrome and obesity have been reported with deletions of chromosome 11p14-p12, a region that encompasses BDNF, the disruption of which is known to cause obesity.42
Summary
The past decade has seen our understanding of the complexity and heterogeneity of inherited syndromes of obesity grow at a rapid pace. These discoveries have had an important, clinically relevant impact on the diagnosis and management of obese patients. First, the genes that underlie most of the pleiotropic developmental obesity syndromes have now been identified—an accomplishment that will greatly facilitate genetic counseling. Second, as exemplified by melanocortin 4 receptor deficiency, we now know that apparently “simple obesity” can be caused by specific genetic defects that influence control of appetite in the hypothalamus. With this realization, we should not assume that “simple obesity,” particularly when it commences during childhood, has simple and readily remediable environmental causes. Finally, as exemplified by congenital leptin deficiency, we now are entering an era when specific molecular diagnoses may lead to specific, effective therapies for inherited subtypes of obesity.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


