Endothelium: Angiogenesis and the Regulation of Hemostasis
Endothelium: Angiogenesis and the Regulation of Hemostasis
Paul J. Shami
George M. Rodgers
NORMAL ANGIOGENESIS
Blood circulation requires the production and maintenance of a vast network of vessels that have specialized functions depending on their organ location. The vascular network involves a complex interaction between endothelial cells (ECs), specialized cells such as smooth muscle cells and pericytes, and the extracellular matrix. Vasculogenesis is the de novo development of vessels.1 It is seen mainly at the embryonic stage of development with the differentiation of a common pluripotent precursor, the hemangioblast, into endothelial and hematopoietic cells. Angiogenesis is the development of new vessels from pre-existing vessels.1 It is an essential process for wound healing and the maintenance of the integrity of the vascular network. Pathologic angiogenesis is seen in disease states including cancer, retinal, and autoimmune diseases.2
As outlined by Conway, Collen, and Carmeliet,1 physiologic angiogenesis is a well-organized stepwise process that involves dilation and increased permeability of the parent vessel, dissolution of the extracellular matrix, division and migration of EC, cord formation, and the development of lumina, and, finally, the maintenance of new vessel integrity. The entire process involves the complex and choreographed effects of multiple inducers and inhibitors (Table 19.1).
The first step in angiogenesis is vasodilation. This is mediated through the activation of the soluble guanylate cyclase by nitric oxide (NO).3 NO also up-regulates vascular endothelial growth factor (VEGF) production.4 By causing intercellular adhesion molecules to redistribute (platelet-EC adhesion molecule-1 and VE-cadherin, among others), VEGF induces an increase in vascular permeability.5,6 The VEGF-induced increase in vascular permeability is negatively controlled by angiopoietin-1 (Ang1) through its receptor, Tie2.7 The next key step to vascular development is the dissolution of the extracellular matrix, which is accomplished by proteases belonging to the matrix metalloproteinase family.8,9 These proteases also induce the liberation of EC growth factors from the extracellular matrix, including VEGF and basic fibroblast growth factor. The action of matrix metalloproteinases is negatively controlled by a family of protease inhibitors, including the tissue inhibitors of metalloproteinases.10
Degradation of the extravascular matrix allows the development of the key element of the angiogenesis process, namely, EC division and migration. The list of factors that stimulate this process is extensive (Table 19.1), but a key role is played by VEGF in concert with Ang1.6,11,12,13,14,15 and 16 Angiopoietin-2 (Ang2) could have angiogenic effects in the presence of VEGF, whereas it is antiangiogenic in the absence of VEGF.14,15,16 and 17 The role played by the endothelial NO synthase and NO has been the subject of controversy, with reports showing that NO has both pro- and antiangiogenic effects.18,19,20,21 Other factors that stimulate angiogenesis include basic fibroblast growth factor and platelet-derived growth factor.22,23,24 EC growth is negatively controlled by endogenous angiogenesis inhibitors that include angiostatin, endostatin, interferons, and antithrombin.25,26,–28 ECs then migrate in large part through the action of integrins (αVβ3 and αVβ1).29 The end result of EC division and migration is sprouting and the formation of cords.1 This is followed by lumen formation, which is controlled by different VEGF isoforms, Ang1, and integrins.1,15 Thrombospondin-1 acts as an endogenous inhibitor of lumen development.1
Once formed, new vessels survive for years.1 This prolonged survival is maintained by the interaction of VEGF with its receptor VEGFR-2, phosphoinositide 3-kinase, β-catenin, and VE-cadherin.1,30 The angiopoietins also play a role in maintaining vessel survival through their receptors Tie1 and Tie2. Ang1 stabilizes the vessel, whereas Ang2 has an opposite effect14,15,31 (see later). An essential element in the maintenance of the integrity of vessels is their “coating” with smooth muscle cells and pericytes.32 Evidence suggests that vascular smooth muscle cells and ECs have a common precursor.33 On stimulation with platelet-derived growth factor-BB, these precursor cells differentiate into smooth muscle cells, whereas VEGF stimulation drives them to differentiate into EC.33 Besides providing physical support for endothelial vessels, smooth muscle cells and pericytes are a source of factors that are important for the maintenance and control of vascular integrity and function.1,32 The extracellular matrix plays a key role in that respect by being a dynamic storage site for growth factors and proenzymes that are important in vessel function and angiogenesis.1
Vascular Endothelial Growth Factor and Its Receptors
VEGF is the pivotal factor controlling angiogenesis. As such, it is the best-studied angiogenic factor. Several proteins belong to the VEGF family and include VEGF (also known as VEGF-A), VEGF-B, VEGF-C, VEGF-D, and placental growth factor.14 Although VEGF-A is the main angiogenic factor discussed here, VEGF-B seems to play an important role in coronary vascular development.34 VEGF-C is essential for lymphangiogenesis by interacting with the VEGFR-3 receptor.35 The function of VEGF-D has yet to be determined.14
Being the major regulator of angiogenesis, VEGF is a mitogen and survival factor for EC.11,14 As mentioned earlier, it is also a potent inducer of vascular permeability, an essential step in the angiogenic process.1,11,14 It has two well-characterized receptors, VEGFR-1 and VEGFR-2 (also known as Flt-1 and Flk-1/kinase domain receptor, respectively).14 They are both tyrosine kinases. VEGFR-2 is the main effector of a VEGF-induced chemotactic and mitogenic response in EC. VEGFR-2 also mediates the ECs’ permeability effects.14 The role of VEGFR-1 in EC response to VEGF has not been totally elucidated. However, it seems to negatively control the VEGF effects by acting as a decoy.14,36,37 Indeed, mice that have been engineered not to express VEGFR-1 have evidence of excess and disorganized angiogenesis.36 In addition to its role in angiogenesis, VEGF has been shown to be trophic for nerve cells, lung epithelial cells, and cardiac muscle fibers.38
Angiopoietins and Their Receptors
Angiopoietins and Tie receptors play an important role in angiogenesis. To date, four angiopoietins have been identified.14 However, only Ang1 and Ang2 have been fully characterized. They interact with the Tie tyrosine kinase receptors, mainly Tie2. Ang1 plays an important role in stabilizing the vasculature.14 Supportive cells express Ang1 and interact with EC through the Tie2 receptor. Genetically engineered mouse embryos that lack Ang1 develop a normal primary vasculature. However, they do not undergo further vascular remodeling.16 Transgenic mice that overexpress Ang1 have evidence of vascularization characterized by larger vessels rather than a greater number of vessels.15 Additionally, those vessels are resistant to leak, further supporting the role of Ang1 as a stabilizing factor.
TABLE 19.1 ACTIVATORS AND INHIBITORS OF ANGIOGENESIS
Adapted from Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res 2001;49:507-521.
The function of Ang2 has been more difficult to characterize.14,17 It too binds with high affinity to the Tie2 receptor. Transgenic overexpression of Ang2 in mice is embryonically lethal and induces a phenotype that is similar to Ang1 or Tie2 knockout experiments. Thus, it has been suggested that Ang2, by acting as an antagonist of Tie2, negates the stabilizing effects of Ang1 on the vasculature. As such, Ang2 may be a destabilizing factor that helps initiate angiogenesis and vascular remodeling.14
NOTCH Signaling
Carmeliet and Jain have proposed a model for vessel development whereby in the angiogenesis process, ECs can be divided into two categories, namely tip cells and stalk cells.39 Tip cells migrate and lead vessel development whereas stalk cells divide. The process is controlled by the NOTCH signaling pathway. VEGF activation of VEGFR-2 leads to up-regulation of DLL4 in tip cells. DLL4 then activates NOTCH in stalk cells. NOTCH down-regulates VEGFR-2 and up-regulates VEGFR-1 in stalk cells, making them less sensitive to VEGF-stimulated sprouting. The end result is to maintain the lead of tip cells in vessel development.40 However, NOTCH stimulates stalk cell proliferation in vivo through activation of WNT signaling.41 NOTCH up-regulates its own inhibitor Nrarp in stalk cells.42 It has been observed that the tip cell position is dynamic with stalk cells moving into the tip position depending on modulation of VEGFR-1 and VEGFR-2 expression.39,42
Origin of Endothelium
Asahara et al. have shown that human buffy coat cells can differentiate into cells expressing endothelial markers, including VEGFR-1, VEGFR-2, and CD31.43 This raises the possibility that circulating endothelial stem cells can be recruited to sites of angiogenesis. This may be particularly relevant for tumor angiogenesis, whereby tumors can develop their vasculature both from recruitment of local endothelium and circulating endothelial stem cells.44 Factors involved in the recruitment of endothelial stem cells may include stromal cell-derived factor-1, thrombopoietin, and soluble kit ligand.44
Angiogenesis in Normal and Malignant Hematopoiesis
There is mounting evidence suggesting the presence of a common precursor for ECs and hematopoietic cells.45 This hemangioblast gives rise to both ECs and hematopoietic cells in embryonic development. Embryonic stem cells express VEGFR-2 and can give rise, depending on culture conditions, to hematopoietic progenitor cells and angioblasts.45 Stimulation of hematopoietic stem cells with growth factors, including kit ligand, interleukin-3, granulocytemacrophage colony-stimulating factor, and granulocyte colonystimulating factor, induces the release by those cells of VEGF, which then induces the release of hematopoietic growth factors by bone marrow ECs.45 Thus, there is a dynamic interaction between hematopoietic and endothelial elements in the bone marrow. This interaction seems to modulate, at least in part, hematopoiesis.
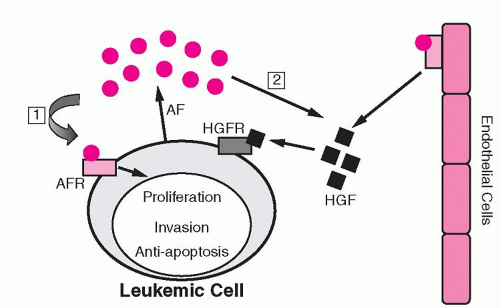
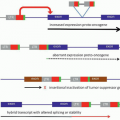
Several studies have shown evidence of increased angiogenesis in hematopoietic malignancies.46 Such evidence has been demonstrated in multiple myeloma and lymphomas, as well as in acute and chronic leukemias.46,47,48,49,50,51,52 and 53,55,56 Malignant hematopoietic cells have been shown to produce angiogenic factors, including VEGF.46 Expression of angiogenic factors has been suggested to have prognostic significance in hematopoietic malignancies, although results have been variable.46 Similar to the effect observed in normal hematopoiesis, VEGF stimulates the production of hematopoietic growth factors by ECs.46 Consequently, malignant cells exploit their environment to their advantage by developing a synergistic relationship with ECs (Fig. 19.1). This has led to the active investigation of antiangiogenic agents as a novel therapeutic strategy for hematologic malignancies.
In addition to the effects of vascular endothelium in modulating and responding to angiogenic stimuli, vascular endothelium also influences other functions,56,57 including vasoconstriction, selective permeability, hemostasis, antigen presentation, and the inflammatory response. The EC surface is a dynamic interface between soluble and cellular constituents of the blood and the remainder of the body.58 A brief discussion of ECs structure and regulation of hemostasis follows.
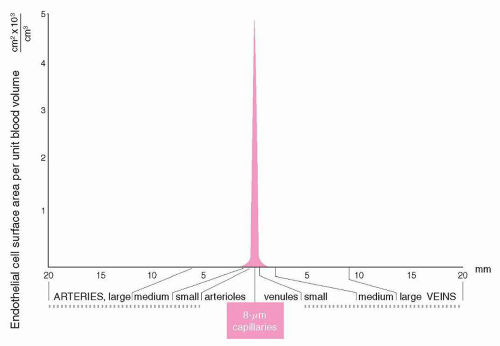
FIGURE 19.2. Relationship of vascular surface area to volume in the vascular system. Diameters are plotted from published data. (From Rushmer RF. Properties of the vascular system. In: Cardiovascular dynamics. Philadelphia, PA: WB Saunders, 1976. Modified from reference 59, with permission.)
FIGURE 19.1. Hypothesis for the role of angiogenesis in leukemia. (1) Angiogenic factors (AFs) produced by leukemic cells can stimulate cell growth and invasion, or inhibit apoptosis (autocrine mechanism). (2) AFs produced by leukemic cells can also stimulate endothelial cell proliferation and the production of endothelial cell hematopoietic growth factors (HGFs) (paracrine mechanism). AFR, angiogenic factor receptor; HGFR, hematopoietic growth factor receptor. (From Dickson DJ, Shami PJ. Angiogenesis in acute and chronic leukemias. Leuk Lymphoma 2001;42:847-853, with permission.)
ENDOTHELIAL CELL STRUCTURE
Individual ECs measure approximately 20 to 50 µm2 in surface area. The total vascular surface area in a normal adult is estimated to be at least 4,000 m2.58 However, the geometry of the vascular system is not static. As indicated in Figure 19.2, the surface area facing a unit volume of blood differs, depending on the vascular bed being considered. For example, the surface areato-volume ratio is approximately 1,000 times greater in capillaries than in large blood vessels.59 This vascular geometry has implications for regulation of hemostasis and is discussed later.
ECs are anchored to the vessel wall by basement membrane secreted by ECs and smooth muscle cells. Basement membrane contains a large number of connective tissue components, including collagen, microfibrils, glycosaminoglycans (GAGs), fibronectin, and thrombospondin. These components may serve as ligands for a number of cell adhesion processes that are important in angiogenesis, hemostasis, vascular repair, and inflammation.57,60
ECs typically exist as a cell monolayer, exhibiting contact inhibition and a cobblestone appearance (Fig. 19.3). Two types of cell-cell junctional structures have been reported: adherens junctions and tight junctions. These structures regulate permeability and maintain polarity.61 Two cell receptors thought to be important in EC monolayer organization are platelet-EC adhesion molecule-162 and vascular cadherin.63 CD146 (MelCAM) is associated with the EC cytoskeleton and likely serves as an EC junction component.64 Members of the junctional adhesion molecule family appear to be components of tight junctions.65
ECs contain unique intracellular structures called Weibel-Palade bodies66; these organelles contain the adhesion protein von Willebrand factor, which is secreted constitutively and also in response to cell stimulation.67 The Weibel-Palade body membrane contains P-selectin, which is expressed on the EC surface after EC activation. When expressed on the vascular surface, P-selectin mediates neutrophil and monocyte adhesion to the vessel wall.68 Selectin-independent platelet adhesion to endothelium has also been reported.69 The role of P-selectin in inflammation and thrombosis is being increasingly recognized.70 Integrins mediating platelet-EC and leukocyte-EC interactions are discussed in Chapters 17 and 7, respectively. Additional EC proteins have been reported to undergo regulated release or cell-surface expression, including tissue plasminogen activator (TPA), interleukin-8, endothelin-1, and multimerin.71 These and other proteins may be contained in Weibel-Palade bodies or other distinct organelles. The regulated secretion of these EC proteins has been reviewed.71
Only gold members can continue reading. Log In or Register to continue