Effector Mechanisms in Immunity
Frixos Paraskevas
MAJOR HISTOCOMPATIBILITY COMPLEX
The function of the immune system is under genetic control1 and molecular genetic studies have helped to elucidate the mechanism of gene regulation of immune functions.2 The genes that regulate immune functions are found in the chromosomal region of the major histocompatibility complex (MHC) involved in defining histocom-patibility for transplantation, and these antigens have been given specific names in different species, i.e., human leukocyte antigens (HLAs) in humans and histocompatibility-2 or H2 in the mouse.
The HLA complex extends approximately over 4,000 kilobases (kb) on the short arm of chromosome 6, i.e., 6p21-3, whereas the H2 complex is located on chromosome 17 and occupies a segment that is halfway between the centromere and the telomere. Approximately 180 genes have been located within the HLA region and the number grows constantly with the increase in sensitivity and sophistication of techniques of molecular biology.
The complex is divided into three regions: (a) the class I region occupies the most distal (telomeric) end of the short arm of the chromosome and spans approximately 1.8 megabases (Mb); (b) the class II region (approximately 1 Mb) occupies the most centromeric end of the chromosome; and (c) the class III region is between the other two regions2 (Fig. 14.1).
Class I Region
Of the polymorphic class I genes HLA-A, HLA-B, and HLA-C, the HLA-B is the most centromeric and HLA-A is the most telomeric; the HLA-C is between the two but closest to HLA-B. There are several nonclassical class I or Ib genes that include HLA-E, HLA-F, HLA-G, HLA-H, HLA-I, HLA-J, HLA-K, and HLA-L, as well as several pseudogenes and gene fragments.2,3,4,5 Pseudogenes give products that do not associate with β2 microglobulin (β2m), probably as a result of deleterious mutations, and the gene fragments are not expressed because the genes are severely disrupted. In the human MHC class I region, which spans 1.8 Mb from the MICB gene to the HLA-F gene at the telomeric end of the HLA region, there are 118 genes (73 known and 45 new genes), i.e., one gene for every 15.2 kb. The G+C content on average is 45.8%6 and contains three classical HLA class I genes, HLA-A, HLA-B, and HLA-C; three nonclassical class I genes, HLA-E, HLA-F, and HLA-G; and two of the nonclassical MHC class I chain-related (MIC) genes MICA and MICB. The class I region also contains 50 non-HLA genes. A number of diseases including Behçet syndrome, ankylosing spondylitis, ulcerative colitis, Takayasu arteritis, and Hashimoto thyroiditis, are associated with a particular class I allele. The large-scale genomic sequencing has greatly facilitated the clarification of gene organization and identification, as well as mapping of the disease-susceptible genes.7 A total of 758 microsatellite repeats have been identified consisting of two, three, four, or five nucleotides. They can be used as markers for mapping the exact location of the disease that is associated with the HLA genes. Class I genes are composed of eight coding sequences or exons that are separated by noncoding sequences or introns. Of the eight exons that correspond to the domains of the protein the first encodes the hydrophobic N-terminal precursor or leader sequence and the second, third, and fourth exons encode the three extracellular domains. A fifth exon encodes the hydrophobic transmembrane segment and the basic residues that act as an anchor. The sixth and seventh exons are small, coding for the cytoplasmic domain of the molecule. The second and third exons are the most polymorphic whereas the fourth exon is the most conserved.
Class II Region
The MHC class II molecule consist of two chains, α and β, each having two extracellular domains α1, α2 and β1, β2 and a transmembrane domain, which anchors the molecule to the cell membrane.5 A peptide-binding groove formed by the α1 and β1 domains of the molecule presents the foreign peptides to CD4+ lymphocytes. The nonclassic molecules are detected in internal membranes such as lysosomes. The human class II molecules were originally defined by mixed lymphocyte cultures and because participation of class I, HLA-A, HLA-B, and HLA-C was excluded, the new region was called HLA-D and its specificities were defined with serological reagents obtained from multiparous women. The HLA-D (DR) is located on the centromeric (left) side of the HLA-B and consists of three subregions which in the direction of the telomere are HLA-DP, HLA-DQ, HLA-DR. The genes of the class II region have a 40% G, C content with only one gene for every 25 kb, i.e., much lower in density than class I (15.2/kb) or class III (14.3/kb) regions. The α chain is composed of five exons and the β chain consists of six exons. The sequence of the α chain contains five regions: i.e., (a) signal sequence, (b) α1 domain, (c) α2 domain, (d) transmembrane region and cytoplasmic tail, and (e) an untranslated 3′ region. By comparison with murine class II genes, considerable gene expansion has apparently taken place in the evolution of human MHC. Many more α and β chains are in the human class II region and the DR molecule is homologous to the murine E molecule whereas the human DQ molecule is homologous to the murine A molecule.
The genes encoding the α and β chains of class II molecule are designated A and B, respectively, and a DR contains one A gene, three B genes, and pseudogenes.8 The α chain can associate with two different β chains to give two different molecules. In the DQ and DP subregions, only one set of genes (A and B) is expressed, and the genes in the other set are pseudogenes. Not all class II molecules that are encoded by these genes are polymorphic. For example, the DRα chain is not polymorphic, and the DPα shows only limited polymorphism.
Within the class II region, several genes encode proteins that are related to antigen processing. Two of them, PSMB9 and PSMB8 (previously known as LMP2 and LMP (for large multifunctional protease), are subunits of the proteasome that cleaves peptides from antigens that are present in the cytoplasm to be transported and loaded on class I molecules.9
Also close to these genes are the TAP1 and TAP2 genes (transporters associated with antigen presentation), which encode the proteins that transport the peptides from cytosol across the endoplasmic reticulum (ER) membrane to be loaded on class I.10 The DR subregion has several haplotypes. The DRA gene is the most telomeric of all class II genes.
The DQ subregion has two pairs of DQ genes, but one of them is nonfunctional. The DP also has two pairs of genes, but only one is expressed, and the others are pseudogenes. Within the class II region are the genes for the nonclassical molecules DMA and DMB, which are distantly related to classical loci (approximately 30% amino acid identity) and must have duplicated from them at an early stage. There are no polymorphic variations in humans in the DM gene. The DN and DO genes, as does DM, may play a role in peptide editing.11
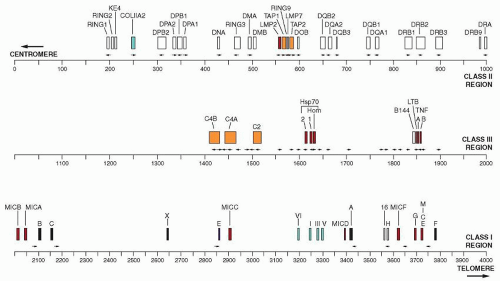 FIGURE 14.1. The human major histocompatibility complex contains genes that are grouped in three regions. Closest to the centromere is the class II region, followed by the class III region, and finally the class I region. Within the class II region are proteasome genes and TAP-1 and TAP-2 genes. The class II genes are located from the centromere in the order DP, DQ, and DR. The class III region contains genes of the complement system heat-shock protein 70, tumor necrosis factor-α, and tumor necrosis factor-β. The class I region contains the genes for the α chain of the HLA-A, HLA-B, and HLA-C molecules and the genes that encode the newly discovered class Ib molecules, such as HLA-E, HLA-F, HLA-G, HLA-H. |
Other genes in the class II region are antigen-processing genes. They constitute a cluster of four genes. Two of them, TAP1 and TAP2, are adenosine triphosphate (ATP)-binding cassette (ABC) transporters (for structure and function, see section “Antigen Processing and Presentation”). The proteasome genes, PSMB9 and PSMB8, are both members of the catalytic subunit of proteasome (see section “Antigen Processing and Presentation”).
Class III Region
The class III genes encode proteins that do not participate in antigen presentation, such as the genes of certain components of the classical and alternative complement pathways (C4, C2, and B), as well as the genes of tumor necrosis factor TNFA and TNFB, HSP70 and the 21-hydroxylase-producing gene (CYP21A2). Telomeric to the class III region is a group of genes that encode the proteins involved in inflammation or infection, such as the tumor necrosis factor (TNF)-α, TNF-β (LTA), and LTB cytokines. It has been proposed that this region should be named the class IV region.12
The genes of class I and class II regions are present in multiple alleles or, in other words, alternate forms that are defined by distinct DNA sequences. The combination of all HLA-I and HLA-II alleles is known as the haplotype. Because the HLA genes are expressed co-dominantly, each individual therefore inherits two haplotypes. Each class I locus has several alleles, and, according to an adopted nomenclature, each allele is defined by its locus, an asterisk, and a four-digit number. The number that defines the allele relates to the serologically defined antigen that is detected on a molecule that is synthesized by the allele (first two digits) and allows discrimination between closely related alleles (last two digits). For example, the B27 locus has at least seven identifiable alleles, which are defined as HLA-B*2701 through HLA-B*2707.
Major Histocompatibility Complex Class II Molecules
Class II molecules are encoded by the MHC genes HLA-A, HLA-B, and HLA-C. Another group of molecules that are structurally homologous to the class I molecules (44% to 82%) has been detected. These molecules are known as class I-b13 to distinguish them from the classical class-I molecules that are now called class I-a. Some of the class I-b molecules are encoded by the MHC-linked genes, HLA-E, HLA-F, and HLA-G, whereas others, such as CD1 and certain CTMV genes, are encoded by non-MHC genes. The MHC class II molecule is a heterodimer consisting of an alpha (α) and a beta (β) chain both encoded by the MHC, whereas the class-I molecule is a heterodimer consisting of an α-chain and the β2m of 12 kD14 (Fig. 14.2). The α chain is divided into three extracellular domains, α1, α2, and α3 Except for the domain that is most distal to the membrane (α1), the other two consist of a loop that is formed by a disulfide bond. The transmembrane portion consists of hydrophobic residues and terminates in five amino acids, including three arginines (Arg) and one lysine (Lys) that anchor the molecule to the membrane. The variable residues are clustered in seven sequences,15 three of which are in the α1 domain and four of which are in the α2 domain. The α3 domain is highly conserved. Amino acids 223, 227, and 229 in the α3 domain are critical for interaction with CD8.16
The structure of β2m consists of one Ig domain with one disulfide bond and is highly homologous between species and homologous to the CH3 domain of human Ig and to the α3 domain of the class I molecule to which it is noncovalently associated.
The L chain plays an important role in the transport of the α chain, because cells that are derived from patients with Burkitt lymphoma, who are unable to synthesize β2m, do not express
HLA antigens. All three extracellular domains of the α chain are in contact with β2m.17
HLA antigens. All three extracellular domains of the α chain are in contact with β2m.17
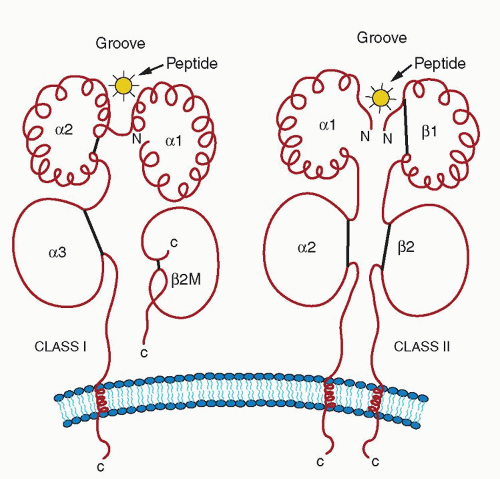 FIGURE 14.2. Structure of class I and class II major histocompatibility complex molecules. The class I molecule is a heterodimer of a heavy chain or α chain and a noncovalently associated light chain, β2 microglobulin (β2m). The α chain consists of three domains: the N terminal α1, followed by α2 and an immunoglobulinlike α3 domain. The β2m protein is homologous to the CH3 immunoglobulin domain, located underneath the α1 domain. The class II molecule is a heterodimer of two chains, α and β. The N-terminal polymorphic domain from each chain forms the peptide groove. |
β2m is present on all cells, and small amounts are shed into body fluids. Because of its low molecular weight, it is filtered out rapidly by the glomeruli, but it is re-absorbed from the kidney tubules and then catabolized. In patients with kidney diseases in whom tubular function is impaired, increased amounts of β2m are detected in the urine. It is also found in increased quantities in the serum and in other fluids from individuals with certain malignancies, such as lymphomas and myelomas.
The crystallographic structure of HLA class I markedly helped the understanding of the function of the molecule.18 β2m and the α3 domain form the base that supports the α1 and α2 domains. Each of the α1 and α2 domains consists of four antiparallel β strands and one α helix. This structure is similar to that of the class II molecule shown in Figure 14.3. Seeing them from the top, the way the T-cell receptor (TCR) does, the juxtaposition of the two domains isolates a longitudinal cavity, which is commonly known as the groove, which is 30 Å long and 12 Å wide in the middle. Its floor consists of the β-pleated sheets of the α1 and α2 domains (“intramolecular dimer”), and the side walls are two α helices, one each from the α1 and α2 domains. The MHC I groove tapers at both ends to a width of approximately 5 Å and then is blocked completely by bulky amino acid side chains (Tyr84 and Trp167), which are conserved in virtually all class I MHCs.17 It is lined by a series of pockets (A to F), which are sites of interaction with the peptide.
The polymorphism of the class I molecule is particularly concentrated in the groove,19 and yet certain residues are strongly conserved among virtually all class I molecules. These residues surround, on the left and right ends of the groove, the first and last pockets (A and F), which bind the N terminus and C terminus of the peptide, respectively. The other pockets line the central portion of the groove and are also involved in peptide binding.20 Many of the MHC side chains that contribute to the formation of the pockets are polymorphic, and, as a result, the architecture of the pockets varies among the class I molecules. In some molecules, a bulky side chain may block a pocket that is open in another molecule. In other molecules, a smaller polymorphic amino acid may extend the size of the pocket.21 This may explain the fact that there are sequence restrictions on the final residue of the peptides.22 A given groove can bind hundreds or even thousands of different peptides, which are identical or homologous at only a few side-chain positions.22,23 Side chains from the MHC residues protrude into the groove from the α helices and the β sheet and form an irregular surface along the length of the groove, so that side pockets extend out from the main cavity.24
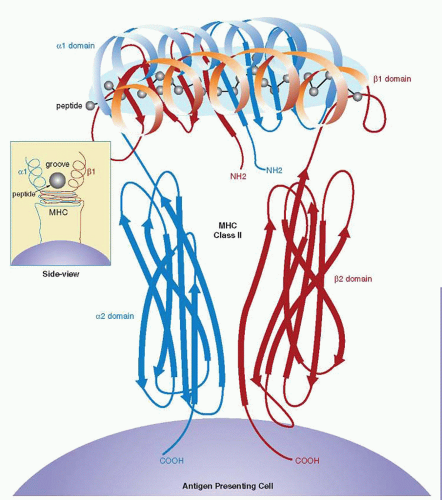 FIGURE 14.3. Drawing of a major histocompatibility complex (MHC) class II moleculepeptide complex. The MCH class II molecule forms a groove with walls that are made by α helices from the α1 and β1 domains and a floor by β-pleated sheets formed by antiparallel β strands from each chain. The peptide is usually long (15 to 25 residues) and projects from both ends, which are not closed (as with MHC class I molecules). |
A class I molecule usually accommodates within the groove peptides that are nine amino acids long and that bind with an affinity that is 100- to 1,000-fold higher than longer or shorter peptides. The binding of the peptides is mediated by certain amino acids that are known as anchor residues (sequence-dependent binding). The set of anchor residues that binds to a certain MHC molecule is known as a sequence motif. It allows the identification of peptides within a complex immunogen that are likely to bind to class I MHC. Hydrogen bonds between the peptide and conserved MHC I side chains provide stability to the peptide binding, which is sequence-independent.25 A prominent kink or arch is found approximately one third of the way from the N terminus, which lifts the peptide up, away from the main floor.
The arch that is formed by the peptide contributes to the antigenic specificity of the complex, despite the fact that 73% to 83% of the peptide surface is buried by the MHC in the complex. A number of side chains that do not affect the binding of the peptide to the MHC contribute strikingly to the TCR binding (flag side chains).26 Antigenic specificity is provided not only by the flag side chains, but also by the conformational flexibility of the
peptide. Peptides that differ in length are usually accommodated by buckling at the center of the cleft.
peptide. Peptides that differ in length are usually accommodated by buckling at the center of the cleft.
In many different peptides, the flag side chains are prominently exposed at the surface of the complex and provide TCR with a direct access to the antigenic identity of the bound peptide. The upward-pointing orientation of flag side chains, such as P1 and P4, is conserved in many different HLA-A2-peptide complexes. Some peptides are sandwiched between the MHC and the TCR and make contact with both molecules.
In conclusion, the requirements to satisfy the vast and exquisite specificity of the TCR are provided by the peptide-MHC complex, which is formed with only a small number of nonvariable MHC molecules. The peptide binding is contributed to peptide-MHC complex formation by sequence-dependent binding and is supplemented by sequence-independent interactions of the primary anchor side chains in specific MHC pockets. These interactions, although constraining the conformation of class I-bound peptides at both ends, leave the center relatively free to adopt a conformation that optimizes the secondary anchor and other interactions with the MHC molecule. The MHC molecule is a remarkable tool with which evolution has achieved so much by so little.
Class II Molecules
The second group of MHC genes, the class II genes, encode glycoproteins that regulate cell interactions in the immune response. The genes reside in the D region of the MHC in humans and in the I region of the MHC in the mouse. The class II proteins are also referred to as Ia antigens (immune response antigens). They are not as widely distributed as the class I molecules. They are found in high concentrations on B lymphocytes and on a subpopulation of macrophages, as well as on dendritic cells (DCs). On T cells, small quantities are detectable under conditions that are associated with T-cell activation in vitro or during the course of certain diseases. However, human T cells may acquire Ia antigens from other cells, and murine T lymphocytes acquire Ia antigens passively from macrophages.27 A variety of cell types expresses Ia antigens in response to interferon (IFN)-γ including vascular endothelial cells,28 melanoma cells, and brain astrocytes.
The human class II molecule is a heterodimer of two noncovalently linked proteins, an α chain (34 kDa) and a β chain (29 kDa).29 Each chain can be divided into four domains, two of which form the extracellular region in each chain, and the third and fourth correspond to the transmembrane segment and the intracellular part of the chain, respectively. With the exception of the α1 domain, the other three domains (α2, β1, and β2) are loops that are formed by disulfide bonds. The α2 and β2 domains are homologous to the Ig constant domain. The human DR, DQ, and DP β chains and the DQ α chains are polymorphic, and only the DR α chains and DP α chains seem to be invariant (Ii).
The assembly and expression of class II molecules involves a third chain that is known as Ii (CD74),30 which is encoded by a gene on chromosome 5. Complementary DNA clones have been isolated, and the amino acid sequence indicates that the Ii chain is a type II transmembrane protein; that is, the N terminal is inside the cell. When synthesized in the absence of MHC class II molecules, the Ii chain exists as a trimer or even as a hexamer.31 The complex of the Ii chain with the class II α and β chains is a nonamer that consists of a core of three Ii chains, each one associated with an α and β class II dimer.32
However, a peptide from the N terminal two thirds of the I chain, which is known as a class II associated invariant chain peptide (CLIP), inhibits the binding of the antigenic peptides to the class II molecule.33 The assembly of the class II Ii chain complex occurs in the ER. Class II α/β dimers that are formed in the absence of the Ii chain are poorly expressed on the cell surface and remain in the ER, as they cannot maintain their normal folded structure. In the assembly of multimeric proteins, certain ER-associated proteins stabilize the partially folded subunits, thus preventing aggregation. These proteins are known as chaperons.34
Another ER-resident protein that associates with class II chains is calnexin.35 The class II molecule is transported to the Golgi apparatus and enters the endosomal-lysosomal system, where it is loaded with an antigenic peptide on its way to cell surface expression36 (see section “Processing Antigen for Class II Presentation”).
The polymorphism of the class II molecule is localized in the α1 and β1 domains. HLADR, HLA-DQ, and HLA-DP have three hypervariable regions in their β1 domains, whereas the α1 domain of HLA-DQ has only one hypervariable region.37 The CD4 molecule interacts with amino acids 134 to 148 of the β2 domain.38,39
The crystallographic structure of the HLA-DR1 molecule reveals similarities with the structure of the HLA class I molecule.40 The α1 and β1 domains form the peptide-binding groove (Fig. 14.3). As with the class I molecule, the floor is the β-pleated sheet, and the walls are the α helices from the α and β chains. The first two hypervariable regions are found on the floor, whereas the third hypervariable region is on the α helix of the β chain. The class II groove can accommodate longer peptides (10 to 25 residues), because, in contrast to the class I groove, it is open on both ends.41 Long peptides, which range from 15 to 25 residues, can bind in an extended conformation, projecting out of both ends of the groove. This contrasts with class I peptide binding, in which mostly nonamers bind with extended, but kinked, conformations, and the N and C termini of the peptides are bound to groove pockets. In class I molecules, the residues at the end of the groove are conserved, imposing tight binding for the peptides, but in the class II molecules, conserved residues are located at different intervals along the groove, making the peptide-binding side chain independent. The peptides in class II bind through the central region, and their N- and C-terminal ends protrude from the open ends of the groove.40
Evidence from crystallographic studies indicates that the class II molecules exist as tetramers on the cell surface.42 Therefore, the geometry of the TCR-MHC peptide interaction places the complementarity-determining region (CDR)-3 loops of the Vα and Vβ chains of TCR in contact correspondingly with the N-and C-terminal ends of the peptide, whereas the CDR-1 and CDR-2 loops are in contact with the β1 and α1 domains. Ligand-induced dimerization of receptors is a general feature for cell activation and involves most of the known receptors.
MAJOR HISTOCOMPATIBILITY COMPLEX MOLECULES: NONCLASSICAL
Class I: Human Leukocyte Antigen-E, Human Leukocyte Antigen-F, and Human Leukocyte Antigen-G
The nonclassical class I HLA molecules are HLA-E, HLA-F, and HLA-G, and are also known as class Ib, to distinguish them from class Ia (HLA-A, HLA-B, and HLA-C). They constitute one group of the class Ib molecules, the other being the CD1 molecules, a non-MHC-linked group with genes that are located in chromosome 1. Class Ib molecules show limited polymorphism, and although their tissue expression is low, they play a significant role as molecules of antigen recognition.42,43 They form heterodimers with β2m and have conserved key residues that interact with CD8.
Human Leukocyte Antigen-E
The structure of HLA-E is similar to that of the class Ia molecules,44 with the β2 and CD8 interaction sites being conserved.
The size of the HLA-E groove is structurally similar to that of the classical molecules. H preserves the crucial positions within the groove, of certain residues that determine the preference for binding the peptide. Peptides occupy all the pockets and as a result the HLA-E keeps the peptide tightly bound. The secure fastening of the peptides reinforces the favorable selection of a specific peptide.45
The size of the HLA-E groove is structurally similar to that of the classical molecules. H preserves the crucial positions within the groove, of certain residues that determine the preference for binding the peptide. Peptides occupy all the pockets and as a result the HLA-E keeps the peptide tightly bound. The secure fastening of the peptides reinforces the favorable selection of a specific peptide.45
The leader peptides of class Ia HLA molecules enter the cytosol after they are cleaved from the coding sequence and are then transported to the inside of the ER by the TAP transporter, to be loaded onto HLA-E.46 Detection of HLA-E on the cell surface of CD56+CD16+ natural killer (NK) cells can be achieved accurately with HLA-E tetramers47 (further information regarding HLA-tetramers can be found in Appendix A). The tetramers react with the CD94/NKG2A receptors, and HLA-E is a major ligand for this inhibitory receptor on NK cells48 (see Chapter 13). Recognition of HLA-E by this receptor inhibits activation of NK cells and thus protects cells from being killed by the cytotoxic NK cells.49 Because HLA-E is expressed with the leader peptides of MHC class I molecules, its presence on the cell surface sends signals to surveilling NK cells that there is no “missing self,” that is, an absence of MHC class I molecules, and thus protects cells from being killed by NK cells. Rare CD8+ T cells have been detected that bind HLA-E directly through the TCR.50 Subversion of the protective mechanism that is conferred by HLA-E has been demonstrated with cytomegalovirus (CMV).
The leader peptide of the CMV glycoprotein gpUL40 is the same as the class Ia leader peptides, and, as a result, it binds to HLA-E.51 Although CMV down-regulates MHC class I expression on infected cells, thus rendering them susceptible to lysis by NK cells (“missing self”), it concomitantly up-regulates HLA-E, thus inhibiting lysis. This mechanism represents an escape route for CMV.
Another example of subversion of HLA-E function is the binding of heat-shock protein, HSP60, signal peptide.52 HSP60 is present in all normal cells and in eukaryotic cells and serves as a mitochondrial chaperon. Stress stimuli, such as heat, nutrient deprivation, toxic chemicals, and inflammation, up-regulate HSP60. Binding of the HSP60 signal peptide to HLA-E results in loss of its recognition by the CD94/NKG2A receptor and renders stressed cells susceptible to NK-cell lysis.
Human Leukocyte Antigen-F
The structure of HLA-F is not known in great detail, but some predictions have been made based on the known structure of HLA-E and the sequence analysis of HLA-F.43 HLA-F tetramers stain monocytes and bind to Ig-like transcript (ILT) 2 and ILT4.47,53 ILT molecules, also known as leukocyte Ig-like receptors, are structurally and functionally related. They recognize HLA class I allotypes, rather than individual peptide-MHC complexes, and are expressed on monocytes, macrophages, and DCs. HLA-F is retained intracellularly, empty, but reaches cell surface after acquisition of its ligand.54
Human Leukocyte Antigen-G
HLA-G shows limited polymorphism, restricted tissue distribution, and low levels of expression and therefore belongs to the nonclassical MHC molecules. In the HLA-G gene, all of the regulatory elements of HLA class I genes are missing, thus suggesting that its expression and distribution are uniquely regulated.55 HLA-G is expressed strongly in extravillous cytotrophoblasts, endothelial cells of fetal vessels, and in other tissues, such as the thymus. It is not detected in syncytiotrophoblasts. Messenger RNA (mRNA) of HLA-G is detected in peripheral blood T and B cells, keratinocytes, fetal liver, and other cells.56 Tetrameric complexes of HLA-G bind to monocytes, especially the CD16+ subpopulation, and react with ILT2 and ILT4 receptors, but they do not bind T, B, or NK cells.47,57
The HLA-G gene transcribes six different transcriptional isoforms, membrane bound and soluble forms.56 The complete transmembrane isoform HLA-G1 consists of three extracellular domains, whereas isoforms HLA-G2, HLA-G3, and HLA-G4 have one or two domains.
The peptide-binding pocket residues determine the specificity of the molecule. The isoforms HLA-G2 and HLA-G3 lack the α2 domain and, as a result, do not bind peptides. They also do not bind the CD8 molecule and are therefore unable to present peptides. The HLA-G molecule has an antigen-presenting potential, but, because of the limited diversity of the peptides and its low polymorphism, the function of the molecule is rather restricted. However, it may still be sufficient, considering the limited species of viruses that infect the placenta.56
Other than antigen-presenting functions, HLA-G may be more important for its role during pregnancy. HLA-G exerts inhibitory functions by several mechanisms. First, it binds directly to CD94/NKG2A, which is an inhibitory receptor58,59; second, it binds to receptor KIR2DL4 on all NK cells that inhibit lysis of cells that express HLA-G60; third, the leader peptide of HLA-G binds to HLA-E, which interacts with the inhibitory receptor CD94/NKG2A. HLA-G is strongly expressed in the placenta, and these inhibitory mechanisms are important for the protection of the fetus.
Class II: Human Leukocyte Antigen-DM and Human Leukocyte Antigen-DO
Human Leukocyte Antigen-DM
There are two nonclassical II proteins with their genes within the class II region, HLA-DM, with two genes, HLA-DMA and HLA-DMB on the centromeric site of TAP and PSMB genes. DMA and DMB encode the α and β chains of the DM molecule,61 which is detected in all class II expressing cells and is up-regulated by IFN-γ, together with classical class II molecules. Class II molecules associate with the Ii chain after they enter the ER and form nonameric complexes (αβIi)3. Through the CLIP, the Ii chain prevents class II molecules from binding peptides within the ER (see details in the following discussion). The heterodimer DM is formed in the ER, and, by the time class II molecules pass through the endosomal system on the way to the cell surface, the DM protein has already arrived there by a multivesicular system that is known as the MHC class II compartment (MIIC). It is retained in MIIC by a tyrosine (Tyr)-based targeting motif (YTPL) that is located in the cytoplasmic tail of the β chain. DM transiently binds directly to the HLA class II-CLIP complex62 and induces the release of CLIP, whereas DM remains associated with the class II molecule until the peptide to be loaded has been found and is safely in place within the groove.63
DM rescues empty HLA-II molecules from denaturation and loss of their peptide-binding capacity. Both HLA-II and DM are in an “acidic” environment, and conformational changes of both molecules seem to facilitate their interaction.64
Another important function of the DM molecule after the removal of CLIP and maintenance of an intact three-dimensional structure of the class II molecule is the editing of peptide loading. Editing implies a selection, from a variety of available peptides, of a good peptide, which is defined by the stability of its interactions within the groove.65 Intrinsic stability is determined by multiple hydrogen bonds between the peptide backbone and the conserved residues of the binding groove and by the anchor side chains and the specificity pockets.66 The mechanism of the ultimate release of DM from HLA-II is not clear. It appears that DM binds more stably to an empty HLA-II than to one that contains a peptide.67
Human Leukocyte Antigen-DO
The genes that encode the α and β chains of the DO molecule are similar to the class II genes. The DO proteins have a more
restricted distribution and are detected in B cells, DCs, a melanoma cell line, and cortical and medullary epithelial cells in the thymus.68 Upon completion of its synthesis, DO associates with DM, and the tetramer is transported mainly to lysosomes, in which most of it is localized.69,70 The lysosomes that contain DO also contain class II molecules. Loading of peptides to class II is slower in the presence of DMDO tetramers, suggesting that DO is a negative regulator of DM function,71 and overexpression of DO results in accumulation of MHC class II-CLIP complexes at the cell surface. The regulation of peptide presentation by DO limits only one of the mechanisms of antigen uptake—that is, by free fluid phase—whereas membrane Ig-mediated uptake is augmented.72
restricted distribution and are detected in B cells, DCs, a melanoma cell line, and cortical and medullary epithelial cells in the thymus.68 Upon completion of its synthesis, DO associates with DM, and the tetramer is transported mainly to lysosomes, in which most of it is localized.69,70 The lysosomes that contain DO also contain class II molecules. Loading of peptides to class II is slower in the presence of DMDO tetramers, suggesting that DO is a negative regulator of DM function,71 and overexpression of DO results in accumulation of MHC class II-CLIP complexes at the cell surface. The regulation of peptide presentation by DO limits only one of the mechanisms of antigen uptake—that is, by free fluid phase—whereas membrane Ig-mediated uptake is augmented.72
Major Histocompatibility Complex Class I Chain-Related Genes and Proteins
A distinct family of genes, the last and final to be detected within the MHC class I region, is known as MIC.73 MICC, MICD, and MICE are in close proximity to HLA-E, HLA-A, and HLA-F, respectively. MICE and MICG are between HLA-G and HLA-F, and MICF is centromeric to HLA-G. MICC, MICE, MICF, and MICG are pseudogenes. MICA and MICB are unusually large genes (11 to 13 kb) compared to an average of 3.5 kb for HLAA to HLAG genes. However, their overall genomic structure parallels those of the other MHC genes and of the Ig superfamily genes in general, in which distinct functional domains are encoded by separate exons. The crystal structure of MICA shows that the molecule has a general configuration of a class I molecule, that is, a membrane proximal Ig-like C-type domain, and an α3 domain that, with two distal α1 and α2 domains, together form an eight-stranded antiparallel β-pleated sheet bordered at the edges by two α helices.74 The groove is less spacious than that of class Ia molecules, i.e.,10 Å within the first four β strands and 7 Å across the second four β strands (compared with >18 Å of classical MHC-I and 14.4 Å for CD-1). The most striking finding is that the groove is oriented down toward the membrane,75 so that the whole molecule appears as a wilting flower.76 Another major difference from other MHC molecules is the lack of β2m binding, due to restructuring of the interface of binding, which ablates the interaction.76 The surprising findings about the structure of these proteins were matched by equally surprising findings about their function. The MIC proteins are recognized by the γδ T cell, especially the Vδ-1 cell, but not by the αβ T cells.77
In addition, MICA is the ligand for NKG2-D activating receptor of NK and γδ T cells, triggering cytolytic responses against epithelial tumors that express MIC-A.78 NKG2-D is the most common NK-cell receptor that is known, but it may require a synergistic action by a TCR.79 The polymorphic residues are concentrated in the α2 domain,74 which apparently is re-oriented after binding with the ligand NKG2-D.80 This ligand-induced re-positioning of the MICA molecule brings the α1–α2 platform with the same polymorphic residues in contact with the NKG2-D receptor. This receptor-induced conformational change is probably facilitated by the coil of the α1 to α3 domains.
A number of disease associations have been detected with MIC alleles, such as ankylosing spondylitis, Behçet syndrome, psoriasis vulgaris, and Kawasaki disease. MICA and MICB are up-regulated under stress and in many gastrointestinal tumors. Pancreatic carcinomas express MICs, and their level in the serum of patients is elevated.81 Soluble MIC unpairs NKG2-D-mediated immunity (see Chapter 13); i.e., it interferes with T-γδ cells and NK cells.82 In summary, the MICA and MICB polymorphic HLA-like molecules present several unresolved enigmas. Their distribution is relatively restricted to epithelial and endothelial tissues (not on T or B cells) keratinocytes as well as monocytes83 and their expression is associated with some form of stress. In spite of their polymorphism, they are recognized by γδ T cells in a genetically unrestricted manner.
CD1 System: Genes and Proteins
One of the first monoclonal antibodies, which was made after the discovery of hybridoma technology,84 was specific for a protein that later was given the first number, CD1, when the CD nomenclature was introduced at the First International Workshop on Human Leukocyte Differentiation Antigens, which was held in Paris in 1982.
The CD1 family consists of four proteins, CD1a, CD1b, CD1c, and CD1d that are encoded by genes CD1A, CD1B, CD1C, CD1D, and CD1E, which is not transcribed. The genes are located on chromosome 1q22-23, and the proteins that they encode have significant homology to MHC class I and II proteins. They are divided into group 1, which contains CD1a, CD1b, CD1c, and CD1e and group 2, which contains CD1d.
CD1 molecules represent an ancient family of antigen-presenting molecules, and the fact that they are homologous to the MHC molecules suggests that both evolved from a common ancestor with subsequent diversification. CD1 consists of three extracellular domains, α1, α2, and α3. The α1 domain has minimal, if any, homology to MHC I, whereas with the other two, homology is approximately 35%, which is still far below the average of 70% between different class I proteins.
All CD1 proteins are transmembrane and associate with β2m, which is necessary for folding and membrane expression.85 The crystal structure of mouse CD1d1 shows a remarkable similarity in overall shape to MHC class I proteins.86 The α1 and α2 domains form a groove with β-pleated sheets that is different from that of the MHC molecules, with three channels, A, C, and F′, and a distinct tunnel, which is designated T′.
Another difference between CD1 and MHC is that the CD1 groove is formed by hydrophobic residues, so it is unlikely to form hydrogen bonds with the peptide; furthermore, it is also closed at both ends as well as covered for much of its length.
A complex of CD1b with ligands has been crystallized, and its structure has been solved 87. The total volume of the groove (220 Å3) and the four interconnected channels are occupied by the two alkyl groups necessary for stable ligand binding.
Mycobacterial mycolates, which are large molecules that play a crucial role in the survival of mycobacteria in the intracellular environment, have been the first characterized ligands for CD1b.88 Lipids with three alkyl groups, such as triacylglycerols that are found in atherosclerotic plaques, are presented by CD1, are expressed on macrophages of the atherosclerotic lesions, and activate T cells.89 The role of this mechanism in the formation and evolution of such lesions remains to be determined. The CD1 molecules are antigen-presenting molecules for lipids: i.e., bacterial as well as self-glycosphingolipids.90 The CD1d molecule presents a number of endogenous glycolipids, tumor-derived lipids as well as nonlipid molecules to NKT cells.91 The presentation of these molecules has immunopotentiating effects that might be of some utility for the treatment and prevention in certain disorders. The CD1 molecules mediate T- (or NKT-) cell activation, following antigen presentation by CD1-expressing DCs.92 Endotoxin binding to TLR4 receptor induces CD1 expression on DCs, promoting T-cell activation.92
UL-16-binding Proteins
The UL-16-binding proteins (ULBP) are novel class I-related surface proteins whose identification was based on their ability to bind the UL-16 glycoprotein of the human CMV.93 The family has three members, ULBP1, ULBP2, and ULBP3, and their genes are located on chromosome 6q25 and not within the MHC complex. However, sequence alignments show clearly that the ULBPs belong to the MHC class I family. The ULBP proteins are glycosyl phosphatidyl inositol (GPI)-linked, do not have the α3 domain of class I MHC, and do not associate with β2m. Expression of ULBPs or the MICs on target cells confers susceptibility to NK-cell killing. The ULBP
transduces signals that override the negative signal generated by an inhibitory receptor. ULBP messages are expressed by a variety of cells, tissues, and tumors. The ULBPs are ligands for the homodimeric NKG2-D C-type lectin receptor,94,95 which is expressed not only on NK cells, but also on T cells and activated macrophages.96 The cytoplasmic domain of NKG2-D is short, and, as a result, signaling is delivered by the DAP-10 adaptor protein that is associated with NKG2-D (see Chapter 13). DAP-10 binds to the p85 subunit of phosphoinositide 3-kinase (PI3K) and adaptor Grb2, transmitting downstream signals from NKG2-D through Janus kinase 2 and Akt activation pathways.95 NKG2-D delivers co-stimulatory signals to T cells,79 stimulates proliferation, and induces increased production of cytokines and other activation-associated molecules.94 UL-16 mediates an additional evasive function of the human CTMV by inhibiting MICB expression, because it binds directly to MICB, causing its intracellular retention.97 Metalloproteases mediate proteolytic shedding of ULBP2 from tumor cells, which interferes with NKG2-D-mediated cytotoxicity and thus may facilitate tumor escape from immunosurveillance.98
transduces signals that override the negative signal generated by an inhibitory receptor. ULBP messages are expressed by a variety of cells, tissues, and tumors. The ULBPs are ligands for the homodimeric NKG2-D C-type lectin receptor,94,95 which is expressed not only on NK cells, but also on T cells and activated macrophages.96 The cytoplasmic domain of NKG2-D is short, and, as a result, signaling is delivered by the DAP-10 adaptor protein that is associated with NKG2-D (see Chapter 13). DAP-10 binds to the p85 subunit of phosphoinositide 3-kinase (PI3K) and adaptor Grb2, transmitting downstream signals from NKG2-D through Janus kinase 2 and Akt activation pathways.95 NKG2-D delivers co-stimulatory signals to T cells,79 stimulates proliferation, and induces increased production of cytokines and other activation-associated molecules.94 UL-16 mediates an additional evasive function of the human CTMV by inhibiting MICB expression, because it binds directly to MICB, causing its intracellular retention.97 Metalloproteases mediate proteolytic shedding of ULBP2 from tumor cells, which interferes with NKG2-D-mediated cytotoxicity and thus may facilitate tumor escape from immunosurveillance.98
Furthermore, soluble ULBP proteins down-regulate NKG2-D expression on NK cells, further interfering with NK-cell-mediated attack on cancer cells.99
ORIGINS OF THE MAJOR HISTOCOMPATIBILITY COMPLEX
The human MHC is close to 4 Mb long, with approximately 180 to 200 genes, which traditionally has been divided into three regions: starting from the centromere to the telomere, class II, class III, and class I. During evolution, genetic events, such as insertions, duplications, deletions, inversions, conversions, and translocations, have introduced modifications, but the conserved genes have escaped identification with ancestral chromosomal regions. Genes that arose within a species by duplication are termed paralogous genes, and the chromosomal segment that contains the duplicated genes is termed the paralogous region. In the late 1990s, it was realized that three other chromosomes, 1, 9, and 19, have genes that are organized in the same manner as the MHC.100,101
Some gene families, such as NOTCH and PBX, are represented in all four chromosomal paralogous regions, whereas others are represented in only two or three. Two hypotheses have been proposed for the explanation of these observations: (a) All four paralogous regions arose from large-scale chromosomal duplication102,103; and (b) the paralogous regions arose from independent duplications of genes, which were brought into proximity by selective forces (the functional clustering hypothesis).104 It is possible that chromosomal duplication may have played a primary role, and functional clustering may have played only a minor one.
The hypothesis proposes that the “block duplication” that generated the four paralogous regions took place before the emergence of jawed vertebrates. Because all vertebrates have four paralogous regions, the duplication from one ancestral region must have occurred twice. It is postulated that the first occurred before the emergence of jawless fishes, which have two paralogous groups, and the second occurred before the emergence of jawed vertebrates. In the vertebrates, the gene order within each region is poorly conserved, probably as a result of structural rearrangements over the 500 million years since the second duplication. The hypothesis of “block duplication” draws severe criticism from others, who have data from phylogenetic analyses of individual gene families that are considered to be inconsistent with this hypothesis.105
Class Ia genes cluster with class Ib genes but have risen independently by gene duplication from classical genes.
Furthermore, the polymorphism of MHC molecules is localized within the peptide-binding region. Within this region, the number of nonsynonymous nucleotide substitutions (amino acid-altering) far exceed the synonymous (silent or neutral) substitutions, whereas the opposite is true of the rest of the molecule. Because the synonymous substitutions are almost neutral, it is evident that polymorphism is supported by a form of natural selection, which favors peptide-binding region diversity. Other genomic evidence from Xenopus laevis (frog) indicates that, in this amphibian, there are class I, II, and III genes, which are linked.106 Similarly, in cartilaginous fish (sharks), the most primitive organisms with a MHC, the gene structures and sequence variations are impressively similar to those in humans. Sharks also share with humans the MHC class I and class II linkage107 that is common to all amphibian, bird, and mammalian species.108 The proposed primordial MHC (proto-MHC) consists of the linkage group class I-PSMB9–PSMB8–TAP2–BRD2–RXRB-class I and several genes that are involved in the preparation of class I presentation, that is, PSMB8 and PSMB9 (for proteasome) and TAP2 for peptide transporter. It is likely that, with further genomic sequencing analyses and phylogenetic tree analyses, the nature and composition of the proto-MHC will be determined. For many years, the HLA haplotype has been considered to be the most important genetic marker of susceptibility to many diseases. It has been found that the strongest disease associations are with alleles at multiple loci rather than with individual alleles. The expression of alleles from multiple loci has been called the ancestral haplotype (AH). AHs are conserved genomic sequences that are separated by recombination hotspots. For example, the 8.1 AHs (A1, CW7, B8, CHAQ0, DR3, and DQ2) are associated with multiple immunologic diseases.109,110 No single gene predisposes to all diseases, but different regions of 8.1 AH are associated with diseases such as insulin-dependent diabetes, systemic lupus erythematosus (SLE), gluten-sensitive enteropathy, dermatitis herpetiformis, common variable immunodeficiency and IgA deficiency. The AH 8.1 is also associated with rapid loss of CD4+ T cells and impaired survival after human immunodeficiency virus (HIV) infection. The precise immunologic mechanisms that give rise to such diverse diseases are not completely known. The AH 8.1 also affects the balance of cytokines with low interleukin (IL)-2 and IFN-γ and normal IL-4 production, with a bias toward the T-helper cell (TH) 2 type of immune responses.
ANTIGEN PROCESSING AND PRESENTATION
Antigen-presenting Cells
The development of humoral and cell-mediated immunity depends on complex cellular interactions that involve lymphocytes and nonlymphoid cells. Activation of lymphocytes by antigen requires its processing and presentation by another cell that is commonly known as an antigen-presenting cell (APC; Table 14.1). Macrophages promptly capture foreign substances, and antigen processing and presentation were first demonstrated there. Other APCs are the DCs (see section “Dendritic Cells”) and B lymphocytes.
Processing Antigens for Class I Presentation
Antigen processing begins in the cytoplasm with an ATP-dependent, proteolytic machine that is known as a proteasome, which is highly conserved from yeast to mammals.111,112,113,114,115 Most of the MHC class I presented peptides are generated outside the endosomes and lysosomes, and the energy required is supplied by ATP. The proteolytic subunit of the proteasome is a large cylindric structure of 20S, which is arranged in four stacked rings. Based on structural analysis, the 20S proteasome is composed of two types of sequences, α and β. The two central rings of the 20S proteasome are composed of seven distinct, but homologous, β subunits,
which surround a central chamber in which proteolysis occurs (Fig. 14.4). The other two outer rings are made of α subunits, which form openings from which substrates enter and leave. Isolated subunits have no proteolytic activity, therefore the subunits must assume a precise conformation in the β ring to be active. It has been determined that the proteolytic mechanism depends on the hydroxyl group of the N-terminal threonine (Thr) of the β subunits. This mechanism of proteolysis by the proteasome is distinct from that of any other known protease. The function of the α subunits may be related to the formation of a scaffolding for the self-assembly of the β rings,116 as well as facilitation of the entry of the substrates.
which surround a central chamber in which proteolysis occurs (Fig. 14.4). The other two outer rings are made of α subunits, which form openings from which substrates enter and leave. Isolated subunits have no proteolytic activity, therefore the subunits must assume a precise conformation in the β ring to be active. It has been determined that the proteolytic mechanism depends on the hydroxyl group of the N-terminal threonine (Thr) of the β subunits. This mechanism of proteolysis by the proteasome is distinct from that of any other known protease. The function of the α subunits may be related to the formation of a scaffolding for the self-assembly of the β rings,116 as well as facilitation of the entry of the substrates.
TABLE 14.1 ANTIGEN-PRESENTING CELLS | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||
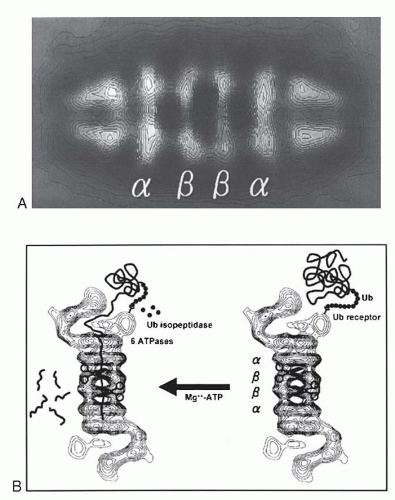 FIGURE 14.4. The proteasome. A: The 26S proteasome consists of a central unit (20S proteasome) composed of seven α and seven β subunits, arranged in four rings in the order αββα. The central unit performs the catalytic functions. On each side of the central cylinder is a regulatory unit. B: Proteins to be degraded are first coupled to ubiquitin (Ub), which binds to an appropriate receptor in the regulatory unit that directs the protein to the central canal (“digestive tract”) of the proteasome. ATP, adenosine triphosphate. (From Tanaka K, Tanahashi N, Tsurumi C, et al. Proteasomes and antigen processing. Adv Immunol 1997;64:1-38, with permission.) |
The 20S (650-kDa) proteasome is capped on each side by a 19S (700-kDa) regulatory complex to form the 26S (2,000-kDa) proteasome113,115 with proteolytic activity. Each 19S cap contains approximately 20 distinct subunits. It has been known as PA700 proteasome activator, because, in the presence of ATP, it associates with the 20S particle and stimulates peptidase activity. In the absence of P700, the 20S particle cannot degrade ubiquitinated proteins. At least six of the P700 subunits are putative ATPases and account for the ATP requirements in proteasome-mediated proteolysis. These ATPases have one or two nucleotide-binding domains (NBDs) (“AAA modules”) and function as chaperons in diverse cellular processes. The size of the 19S regulatory complex, or antechamber, is ˜59 mm3 and is large enough to retain unfolded proteins in a “ready to eat” state before they enter the “execution chamber.”117 The 26S proteasome has a dumbbell shape, with the 20S proteasome as a cylinder in the center and the two PA700 V-shaped subunits attached on each end. IFN-γ induces expression of three β-type subunits, two of them encoded by genes, PSMB8 and PSMB9, within the class II region, and a third one, PSMB10 (MECL1), which is encoded outside the MHC.118 IFN-γ also down-regulates three of the normally constitutively expressed subunits. The net result of INF-γ stimulation is the exchange of three constitutive β subunits by those that are IFN-γ-inducible and that are incorporated into the 20S proteasome.119 This function of INF-γ changes the activity of the immunoproteasome.
Another regulatory complex, PA28 (11S), a 200-kDa conical structure, also forms a cap at either end of the 20S proteasome120 and stimulates multiple peptidase activities with faster kinetics. The PA8 consists of two components, α and β, which are structurally related to the Ki antigen, a nuclear antigen that is detected by autoantibodies in patients with SLE. Ki antigen is now renamed PA28γ. The PA28 at the two ends of the 20S proteasome consists of a heterohexameric complex with alternating α and β subunits. PA28 proteasome is not involved in the initial cleavage of proteins but, rather, degradation of polypeptides of intermediate size. It is thought to work after the initial action of the 26S proteasome.
How from a long polypeptide or a protein, the specific peptide that precisely fits to the groove of the MHC is generated is a tantalizing question. These specific peptides are embedded within the polypeptide chain and in order to be extracted, two precise cuts are needed, one at the N terminal (start) and one at the C terminal (end). The importance of proteasomes for the generation of MHC class I-restricted peptides is widely recognized.121,122
Antigen processing in the cytoplasm does not generate the final product in exactly one step. The view that proteasome makes the C-terminal cut is favored by several studies.123,124 According to this view, the proteasome releases intermediates with a precisely cut C-terminal end but with an extended N-terminal end. Proteasome cleaves peptides on the carboxyl side of basic and hydrophobic residues of substrates. This specificity satisfies an important requirement for the binding of peptide to the MHC class I groove; that is, basic and hydrophobic residues normally serve in anchoring the peptide in the groove. In addition, the C terminally cut peptides are also more efficiently transported by the TAP transporter.
N-terminal trimming is necessary to produce the antigenic peptide and may occur by aminopeptidases in the ER.125 It is estimated that the 26S proteasome generates an antigenic peptide from an intact protein with an efficiency of one peptide for every 20 molecules that are degraded.126 The peptides generated are 3 to 22 residues long, but only 20% are in the range that is favored for high-affinity binding (8 to 10 residues).127 The crystal structure of the proteasome has revealed that the distance between active Thr residues of adjacent β subunits is approximately 28 Å. This distance determines the length of the generated peptides.
The ubiquitin proteolytic pathway has a crucial role in the degradation of short-lived and regulatory proteins that are important in a variety of cellular processes. Proteolytic cleavage by the proteasome requires the conjugation of the substrate by ubiquitin.128,129 The ubiquitin (8.5 kDa) is one of the most conserved proteins in evolution and is activated to a high-energy thioester bond at its C terminal by an activating enzyme, E1. After activation, one of several E2 enzymes (ubiquitin carrier proteins) transfers ubiquitin to an E3 protein (ubiquitin protein ligase) to which the substrate protein is specifically bound. This E3 protein covalently attaches ubiquitin to the substrate, at which it binds to ε-NH2 groups of an internal Lys residue. This step is repeated until a polyubiquitin chain is formed, which serves as a recognition marker to the 26S proteasome. After degradation, ubiquitin molecules are released to participate in another cycle of proteolysis.130 Substrates linked to ubiquitin may have different fates within the cell, depending on to which residue of the ubiquitin they are attached.131 Substrates linked to lysine 48 (K48) usually are directed for proteolysis, whereas K63-linked proteins signal several other, nonproteolytic intracellular pathways, such as kinase activation, protein trafficking, ribosomal protein synthesis, and others.
Rescue of proteins from degradation is mediated by de-ubiquitinating proteases or ubiquitinating specific proteases, the majority of which are cysteine proteases.132 These enzymes remove the (poly)ubiquitin from proteins, before their translocation into the catalytic chamber of the proteasome.133
The proteasome exhibits three distinct peptidase activities that are associated with β subunits. One is chymotrypsinlike because it hydrolyzes peptides after a large hydrophobic residue, a second is trypsinlike because it cleaves after basic residues, and a third hydrolyzes after acidic residues. Occupancy of the chymotryptic site by a peptide substrate allosterically activates cleavages by the postacidic site, which in turn inhibits the chymotryptic site. By these cycles of activation-inhibition, the proteins are cleaved in pieces and are propagated for further fragmentation by other active sites.
Peptide Transport
Peptides that are generated by the proteasome must be transported across the ER membrane to meet the MHC class I molecules at which they are synthesized (Fig. 14.5).134,135,136 and 137 The first indication that peptides are actively transported to the interior of the ER came from the study of mutant cell lines that had low MHC class I expression and could not present antigen, although their capacity to synthesize the molecules was intact. In the absence of peptides, the class I molecule cannot maintain its quaternary structure, folds up, and is incapable of moving along the constitutive pathway to the cell surface. Two genes have been identified within the MHC complex that encode the transporters of cytosolic peptides.138 They are called transporters associated with antigen presentation (TAPs). Transfection of these genes to the mutant lines that were mentioned previously corrected their defect in antigen presentation. Profound defects in MHC class I-restricted antigen presentation were detected in humans lacking TAP2139 and in mutant mice lacking Tap1.140
The TAP proteins are members of the ABC family that is found in prokaryotes and eukaryotes.141 In mammals, the ABC proteins are grouped into four subfamilies: the glycoproteins, the cystic fibrosis transmembrane conductance regulators, the peroxisomal membrane proteins, and the TAP proteins. From the genomic structure of the human TAP1 and TAP2 genes, it is predicted that the TAP1 protein has 10 transmembrane-spanning segments. The sixth segment is joined to the seventh and the eighth is joined to the ninth by hydrophilic cytoplasmic segments.134 The NBD, which is located in the cytoplasm, is large and consists of several modules. Two are involved in nucleotide binding (Walker motifs), and three others are hydrophilic. The direction of transport varies.
Some members (such as P-glycoprotein) transport their substrates away from the NBD, and others transport their substrates toward the NBD. The TAP proteins are functional only as heterodimers and are located in the ER and the cis-Golgi.142 In addition to the ATP-binding site,143 a peptide-binding site is formed by both subunits.144 Peptide binding is ATP-independent, but its transport is ATP-dependent.145 It is of interest that peptides with 7 to 12 residues and a C terminus are transported more efficiently, and those are precisely the requirements for optimal binding by the class I MHC molecule.146 Because peptides are usually rapidly degraded in the cytoplasm, it is suggested that they may be carried from the proteasome to the TAP proteins by chaperon proteins, such as those of the HSP70 family. Genes that encode such proteins have been found in the MHC region.147 Peptide binding to TAP precedes its translocation across the ER membrane, and TAP acts as a peptide channel permitting the diffusion of peptides across the membrane. Most data, however, are consistent with the transporter function, that is, binding followed by translocation. ATP binding releases the peptide as a result of conformational changes of the NBD.
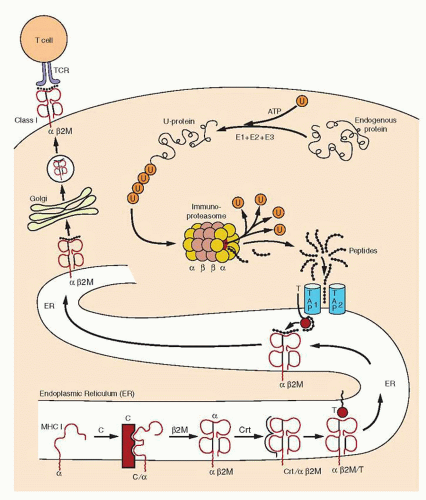 FIGURE 14.5. Processing and presentation for major histocompatibility complex (MHC) class I. Peptides (approximately 8 to 10 amino acids long) are generated by proteolysis within the proteasome. The protein to be degraded is first ubiquitinated. Ubiquitin (U) is attached by a covalent bond to the ε-NH2 group of a lysine residue by an enzyme complex. The peptides are carried across the membrane of the endoplasmic reticulum (ER) by the transporter associated with antigen presentation (TAP) to be loaded on the groove of MHC class I molecules. The nascent α chain of MHC retains its proper conformation by calnexin (C) and after the addition of the β2 microglobulin (β2m) chain by calreticulin (Crt). When the peptide passes inside the ER and lands on the MHC groove, with the help of tapasin (T), calreticulin is relieved from its chaperon duties, because the peptide stabilizes the MHC conformation. ATP, adenosine triphosphate; TCR, T-cell receptor. |
An important question related to class I antigen presentation is the mechanism by which viral proteins or tumor antigens, which are synthesized within the ER, are targeted to the cytosol to be handled by the processing machinery. There is an ER retrograde pathway that transports proteins from the ER to the cytosol.148 Until these proteins are assembled and folded properly, they are retained within the ER by calnexin or calreticulin. Misfolded or aberrant proteins are released for proteasomal degradation.149 However, the ER possesses proteolytic activity, that is, aminopeptidases that may also process N terminally extended precursors of antigenic peptides for loading to MHC class I molecules.
Assembly and Loading of Class I Molecules
The size restriction for peptides to bind to class I molecules is 8 to 10 residues. The restriction is imposed by the structure of the MHC class I groove, which is closed at the two ends. The class I α chain and β2m are synthesized on membrane-associated ribosomes and are inserted co-translationally into the ER through a specialized structure known as the translocon. N-Glycosylation takes place in the ER during the translocation process. HLA-A, HLA-B, and HLAC have a single N-glycosylation site. The ER chaperon, BiP, a HSP70 homolog, interacts with proteins during this stage and, through ATP regulation, provides the energy that is required for the movement of the protein through the ER membrane. BiP binds with free HLA heavy (H) chains.150 Calnexin is the most important chaperon and binds to newly synthesized free class I α chain,151,152 but not to the heterodimer (Fig. 14.5).
Calnexin is a transmembrane lectin and binds to N-linked glycans that bear a single glucose after removal by glycosidases of the extra glucose residues. How calnexin facilitates folding is not known, but the glucose is removed by a glycosidase II and calnexin is released. If the MHC class I molecule has not achieved its proper folding, a second glycosylation cycle follows with another attempt at refolding. Removal of calnexin permits the MHC class I molecule to leave the ER. Another chaperon that has been detected by some studies is calreticulin, which is a soluble homolog of calnexin.153 Both recognize Asn-linked glycans bearing a terminal glucose residue, an intermediate in oligosaccharide maturation present on incompletely folded ER glycoproteins. The successive addition and removal of this glucose residue results in cyclical calnexin or calreticulum interactions with substrate proteins until they are correctly folded and assembled. Calreticulin promotes folding of HLA class I molecules to a state in which at low temperatures they spontaneously acquire peptide-binding capacity.153
The TAP heterodimeric proteins transport the cytosolic peptides that are generated by the proteasome across the ER membrane. They bind to MHC class I molecules through an additional component that is called tapasin (TAP-associated glycoprotein).154 Tapasin is a transmembrane glycoprotein that has an ER retention signal in the cytoplasmic region. The gene that encodes tapasin is in the centromeric end of the human MHC region. Tapasin probably forms a bridge between TAP and MHC class I molecules155 (Fig. 14.5). However, tapasin also stabilizes class I molecules in the absence of peptides by occupying the groove, a function that is performed by the Ii chain in class II MHC molecules. In addition, tapasin is involved in peptide loading156 and also influences the peptide selection.157
An additional molecule that associates with MHC class I molecules at the assembly stage is ERp57, an enzyme that ensures the correct formation of intrachain and interchain disulfide bonds.158 ERp57 is a thiol-dependent reductase and a cysteine (Cys) protease. As a protease, it may play a role in the trimming of peptides, because it remains attached to the complex until the time of the peptide loading.
The order and kinetics of the assembly of the various components of the loading complex is as follows159,160: H chain β2m heterodimers can be recovered within 4 minutes after translation, and the peptide joins 2 minutes later. Shortly after the synthesis of the H chain, one disulfide bond forms in the α2 domain, and a second forms in the α3 domain. Disulfide bond formation is necessary for β2m binding and peptide loading. The β2m is required for physical support of the peptide-binding groove.
Calnexin may be bound to a nascent H chain, but calnexin dissociates as β2m binds to the H chain, and calreticulin is taken up by the heterodimer. The other half of the complex is being assembled in the meantime; that is, TAP forms a complex with tapasin, and both are joined by calnexin and ERp57. When the two halves of the final complex join together, calnexin dissociates, and the final loading complex is complete. Loading of the peptide at this point releases the MHC class I-β2m heterodimer for the secretory pathway to reach the cell membrane. Presentation of certain proteins such as those derived from viruses is presented very rapidly to CD8+ T cells. It was proposed that antigenic peptides originate from defective ribosomal products (DRiP hypothesis)161 from a specialized machinery consisting of immunoribosomes.162
Presentation of Exogenous Antigen by Major Histocompatibility Complex Class I Molecules
The class I MHC system evolved to identify cells that bear peptides that derive mostly from infectious agents. The vast majority of peptides that are presented by MHC I derive from proteins that are synthesized by the cell’s own ribosomes. The proteins or polypeptides that generate these peptides are referred to as endogenous, whereas peptides that derive from proteins or micro-organisms that are phagocytosed are termed exogenous. It has been widely accepted that endogenous antigens trigger a CD8+ class I-restricted response, whereas exogenous antigens activate CD4+ class II-restricted cells. However, several examples over the years have detected class I-restricted CD8+ T-cell responses with dead phagocytosed viruses, bacteria, transplants, dead cells, and others, which were handled by the immune system as exogenous antigens and were targeted to the endocytic system.163 This has been known as cross-priming, which was originally described in mice that were immunized with cells that expressed minor histocompatibility antigens. Today, it is also known as cross-presentation. Hepatitis B surface antigen, given by the exogenous route and submitted to efficient endolysosomal processing, can lead to peptide-loaded MHC class I molecules.164 Sometimes peptides derived from internalized particulate material have access to ER or peripheral phagosomes with membrane contributions from the ER, which then are loaded to MHC class I molecules.165 Cross-presentation also occurs by “immunoproteasomes,” i.e., proteasomes enriched in proteasome proteolytic subunits (PSMB9 and PSMB9) by the action of INF-γ.166 The
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, NOX2, maintains an alkaline milieu of the phagosomes of DC predisposing to peptide presentation rather than protein degradation, which occurs by proteases in the highly acidified lysosomes of neutrophils.
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, NOX2, maintains an alkaline milieu of the phagosomes of DC predisposing to peptide presentation rather than protein degradation, which occurs by proteases in the highly acidified lysosomes of neutrophils.
Cross-presentation by APCs of antigens that are chaperoned by HSP (heat-shock proteins) is described by Shrivastava et al.167 HSPs are highly conserved peptide-binding molecules that control the folding of proteins and prevent their aggregation. HSP70 that is complexed with a variety of synthetic peptides is taken up by APCs and elicits CD8+ T-cell class I-restricted antitumor cytolytic responses.168 The HSP-peptide complexes are internalized by a receptor-mediated mechanism and are subsequently processed by a proteasome and TAP-dependent mechanism or through the endosomal route.169 This is a highly efficient mechanism whereby antigens that follow an exogenous route induce strong CD8+ responses against tumors or viruses.170 Peptides that are introduced into the cytosol induce a strong class I-restricted response only if they are chaperoned by HSP, but not alone.171 Receptors for HSP have been detected on CD11b+ cells,172 and, recently, the receptor for all HSPs was identified to be the CD91 protein173 (see Appendix A). The CD91 is receptor for α2-macroglobulin and, with calreticulin, binds and stimulates uptake of apoptotic cells174; together, they act as receptors for the collectin family and regulate the clearance of organs in the removal of apoptotic cells and cell debris.175 It is postulated that HSP-peptide complexes that are given by the exogenous route might be used in the future to elicit protective immunity against cancer on viruses.176
Although macrophages, DCs, and B cells have been reported to be able to mediate cross-presentation, the evidence collectively suggests that the most important cell is a DC. Interactions between CD4 and CD8 T cells that interact with the same APC are apparently needed.177 Cross-priming is involved, predominantly in response to virus infection, cancer cells, and histoincompatible tissues. Although cross-priming is associated with an immune response, depending on the nature of the antigen (e.g., normal healthy cellular antigens), cross-presentation leads to the opposite of the immune response: cross-tolerance.
Antigen Processing for Class II Presentation
Biosynthesis of Major Histocompatibility Complex Class II Molecules
Expression of class II MHC molecules is restricted mainly to certain cell types, such as macrophages, DCs, and B lymphocytes. However, MHC class II molecules can be induced in other cells by IFN-γ. Regulation of MHC class II molecule expression occurs at the level of transcription. Many factors that regulate their expression are DNA binding and are ubiquitously expressed.178 An important mediator is the MHC class II trans-activator molecule (CIITA), which is not a direct DNA-binding molecule and is detected in cells that express class II molecules constitutively or after induction with IFN-γ. CIITA is essential for the activation of MHC class II promoters that have the SXY regulatory module, i.e., a module consisting of certain motifs known as S, X, and Y boxes,179 as well as for the expression of the Ii chain and HLA DM molecules. The MHC class II molecules and Ii chain are co-translationally inserted into the membrane of the ER through a signal peptide. The Ii is a type II membrane protein: the NH2 terminus is on the cytosolic face of the ER and rapidly forms trimers through interactions of its luminal sequences.180 It also associates with three MHC class II molecules, added sequentially, forming nonamers.36 A central region of the Ii, which is termed CLIP, occupies the peptide-binding region of MHC class II and prevents peptide loading to MHC class II in the ER. Furthermore, the MHC class II-Ii association conceals a motif that retains Ii into the ER, and, as a result, the nonamer complex moves to the Golgi apparatus.181 Leucine (Leu) signals in Ii bind the AP-1 and AP-2 adaptors,182 but more than one sorting mechanism probably exists, depending on the cell type. The MHC class II-Ii complexes are targeted to late endosomes, 183,184 possibly with an intermediate stopover, on the cell membrane or on the early endosome. Permanent localization in the early endosomes (by exchanging the cytoplasmic tail of Ii with that of transferrin receptor) blocks class II presentation, indicating that late endosomes or lysosomes provide the appropriate milieu for class II presentation.185 The CLIP has also been shown to mediate interaction of the Ii chain with MHC class I molecules.186
Generation of Antigenic Peptides for Class II Major Histocompatibility Complex Presentation
Proteolytic breakdown of exogenous antigen in the endocytic pathway is essential for peptide loading. Specific uptake involves endocytosis and phagocytosis, mediated by various receptors in different cells, such as the B-cell receptor (BCR) in B cells and mannose receptors and Fc receptors in macrophages. The mannose receptor recognizes patterns of carbohydrates on the surface of the cell wall of infectious agents, and this increases dramatically the efficiency of class II-mediated presentation by DCs (Fig. 14.6).
The endocytic route that is taken by endocytosed foreign material consists of a complex network of compartments that vary in morphology, physiochemical properties, and content. Three major regions, however, can be distinguished: the early endosomes, with slightly acidic pH, are barely proteolytic; the late endosomes are more acidic and contain some components that are classically used as markers of lysosomal compartments, LAMP1 and LAMP2, and the lysosomes that have a low pH and are rich in hydrolytic enzymes. This route is intersected by another route that originates in the trans-Golgi network (TGN). This route is taken by molecules that come out of the ER, and, depending on specific signals such as dileucine or mannose 6-phosphate, they follow the secretory pathway on the way to the cell membrane or secretion. Foreign antigens that enter the endocytic pathway are subject to gradual proteolysis, which may generate peptides that are suitable for receptive MHC class II molecules.187 Antigens follow different routes in the endocytic pathway, and antigen processing is different. 188 “Exit tracks” from the endocytic pathway join the constitutive secretory pathway for reaching the membrane. Proteolytic activities along the endocytic pathway are acquired progressively.
Acidification is an important factor for protein unfolding, and, as the pH decreases progressively, it influences several steps in the processing of incoming antigens and in peptide loading, such as protease activity, MHC class II molecule aggregation, peptide receptivity, and function of chaperons HLA-DM and HLA-DO.
Antigen processing is carried out primarily by proteases, and most of the lysosomal proteases are known as cathepsins. The final peptide that is suitable for loading onto MHC class II molecules is prepared by the action of the protease alone or with the assistance of the MHC molecule. A protease-guided initial step breaks down large proteins into smaller polypeptide chains, which may be partially accessible to MHC class II molecules. At this point, MHC class II-guided processing allows the protease to trim down unnecessary protrusions outside the binding site.189
Several proteases exist within the endocytic system, endoproteases and exoproteases, and some cooperation between them is anticipated.190 It appears that the first key protease is a Cys endopeptidase that cleaves after Asn residues.191 Once this enzyme takes the first step, other enzymes complete the digestion for the generation of peptides.
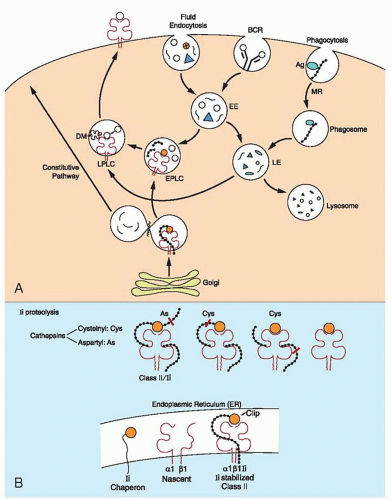 FIGURE 14.6. Processing and presentation for major histocompatibility complex (MHC) class II. Pathogens or their products are captured by phagocytic or dendritic cells and are carried to the interior of the cell enclosed within endocytic vesicles. A: Endocytosis is mediated through the mannose receptor (MR) or the B-cell receptor (BCR), or, depending on the method of entrance, they end up within typical phagosomes or early endosomes (EEs). The endocytic pathway is relatively complex, which is reflected in changes of their composition and pH. There is a dramatic morphologic change from a simple vesicle to multilamellar and multivesicular compartments that are known as MHC class II compartments. The endocytosed material is targeted to vesicles with class II MHC: early peptide loading compartment (EPLC) and late peptide loading compartment (LPLC). Their conformation and groove are maintained by the invariant (Ii) chain in EPLC and HLA-DM in LPLC. The MHC class II molecule is synthesized in the endoplasmic reticulum (ER), and its conformation is stabilized by the chaperon protein Ii chain. A region of Ii termed class II-associated invariant chain peptide (CLIP) occupies the groove and prevents the collapse of MHC class II molecules. MHC class II molecules and Ii exist as nonamers. The complex moves out of the ER, passes through the Golgi apparatus, and reaches the EPLC, where the Ii chain is proteolytically destroyed but CLIP remains in the groove. Exchange of CLIP with the antigenic peptide is catalyzed by the nonclassical HLA-DM protein. In the last step of this long and tortuous path, the MHC class II-peptide complex is delivered to the plasma membrane by the EPLC vesicle, where it interacts with the appropriate T-cell receptor on CD4+ T cells. B: Proteolysis of the Ii chain is mediated by cathepsins, which cleave at a cysteine or aspartic acid residue. LE, late endosome. |
Peptide Loading on the Major Histocompatibility Class II Molecule
As indicated earlier, the MHC class II molecule arrives in the endocytic system as a nonamer with the Ii chain. The CLIP region of Ii is inserted in the peptide-binding groove and prevents interaction with ER-resident proteins or other proteins on the way to the endosomes. CLIP also keeps the MHC class II molecule stable until the time of peptide loading. The precise location of the peptide loading within the endocytic system has been debated for a long time. Electron microscopic studies demonstrate that the MHC class II molecule is found throughout the endosomal pathway, including mannose 6-phosphate-positive late endosomes and in late endosomes and lysosomes, a compartment that is rich in MHC II but negative for the mannose 6-phosphate receptor, which is known as MIIC.192 This compartment consists of vesicles 200 to 300 nm in diameter with heterogeneous morphology, such as multivesicular, multilamellar, and tubulovesicular. Various functional stages of MIICs are distinguished on the basis of the Ii detection; for example, in some of them the intact Ii chain is detected, in others only the aminoterminal region is detected, and, in still others the Ii chain is absent. Loss of the Ii chain is associated with a change from multivesicular to multilamellar MIIC.193 Another endocytic compartment, known as the CIIV (for class II vesicles), from a B-cell line, is similar to MIIC. Both are considered to be sites for peptide loading. The MIIC compartment is set aside from the remaining endosomal vesicles, and several endocytic markers are absent. These vesicles receive newly synthesized MHC class II molecules by virtue of the targeting motif of the Ii chain. Once they arrive, the Ii targeting motif is removed (C-terminal region), and the MHC II-Ii complex is retained within the MIIC for prolonged periods of time.193a The MIIC is particularly abundant in DCs which are the professional APCs.
The first step in the peptide loading is the digestion of the Ii chain in the MHC class II-Ii chain nonamer complex (Fig. 14.6). The carboxyterminal region of Ii is cleaved by a protease that releases an MHC class II-Ii trimer.194 Ii degradation occurs in an orchestrated fashion until only the CLIP remains attached to the MHC class II molecule. Proteolysis proceeds in two stages, and, for proteolysis of the second intermediate, different cathepsins are used in different APCs. Of the lysosomal enzymes, cathepsin S plays an essential role in the degradation of Ii in B cells and DCs, and its absence has major consequences for the onset of humoral immune responses.190 Cathepsin L, on the other hand, is expressed on thymic epithelial cells.195
Removal of CLIP from the peptide-binding groove and subsequent loading with antigenic peptide is catalyzed by the nonclassical class II HLA-DM, which binds directly to MHC class II-CLIP complex,61 near the N terminus of MHC class II.196 It is thought that the DM induces a transitional “open” state to the MHC class II groove that releases CLIP. This occurs by disruption of a few hydrogen bonds between MHC class II and CLIP.197 It is intriguing that HLA-DM does not interact with MHC class II that is associated with intact Ii chain.
The HLA-DO molecule is a negative regulator of HLA-DM, and, when it is overexpressed, it results in an accumulation of MHC-CLIP complexes.
MHC class II-peptide complexes are transported to the cell surface by vesicles, the movement of which depends on the cytoskeletal elements. MIIC transport is microtubule-dependent.198 Microtubules are assembled from αβ tubulin heterodimers in the microtubule organizing center and extend toward the periphery. A large family of motor proteins, known as kinesins, mediate transport of vesicles of the secretory pathway, cytotoxic granules, lysosomes, and the like. On microtubules, kinesin transports vesicles toward the positive end of the microtubule (the end away from the microtubule organizing center, toward the cell periphery). Another group of proteins, dynein, creates motion toward the negative end of the microtubule.199 Thus, dynein is important to keep vesicles around the nucleus, because inhibition of its function shuttles all the vesicles to the cell membrane. How and when the vesicular movement is regulated is unknown. In monocytes, IL10 triggers positioning of MIIC in the cell, but in B cells, the BCR performs this function. The signal for kinesin to transport the mature MIIC—i.e., the MIIC that is loaded with the peptide—to the cell surface is unknown. It is also intriguing that only MHC class II-peptide complexes are detected on the cell surface, not the other contents of the MIIC vesicle.
Antigen Presentation by CD1
CD1 was first identified in human cortical thymocytes but later was detected on other cells, such as on antigen-presenting DCs, and CD1a is expressed on Langerhans cells (LCs), which are specialized DCs in the epidermis.200 CD1c is detected on a subset
of peripheral blood B cells and B cells of the marginal zone, and CD1d is expressed in intestinal epithelia and in a wide variety of hematopoietic cells. The members of the CD1 family are antigen-presenting molecules, and, after their synthesis, they must first reach an appropriate vesicular compartment for peptide loading on the way to the cell surface. There are striking differences in the intracellular distribution of the CD1 isoforms.201,202 CD1a is found mainly on the cell surface but also in early endosomes203,204 and in LCs that are present in the Birbeck granules. Together with CD1d, CD1b accumulates in the late endosomes and lysosomes, whereas CD1c is found in the early endosomes (transferrin receptor-positive). The ratio between surface expression and intracellular concentration also varies among the isoforms. The differential expression of the isoforms is regulated by a Tyr-based motif in the intracellular region that is present in all isoforms except CD1a.205,206 This sequence, YXXZ (Y, Tyr; X, any amino acid; and Z, an amino acid with bulky side chains), interacts with a member of the cytosolic adaptor proteins AP-1 through AP-4.207 AP-2 is positioned in the TGN and mediates delivery from the TGN to the endosomal-lysosomal system, whereas AP-2 is localized in the plasma membrane, where it directs proteins to clathrin-coated pits that lead to the entrance into the endocytic pathway. The AP-3 is implicated in the sorting of proteins from the TGN to endosomes and lysosomes. The AP proteins determine the specificity of destination for the CD1 molecules, but the binding to a specific AP member is determined by the residues that surround the Tyrbased motif.208 The YXXZ motif is essential for localization in the endosomal and lysosomal compartments, because mutations that are introduced into the sequence inhibit presentation of mycobacterial glycolipids to T cells.209 Although the YXXZ motif predicts the final destination, it does not indicate the route that is to be followed. The CD1d first follows a direct route from the ER to the cell surface, where, through the YXXØ motif (Ø, phenylalanine), it binds to AP-2. It is subsequently internalized and is directed to the MIIC compartment.209 MIIC overlaps considerably with lysosomes and may represent a fusion product between endosomes and lysosomes. CD1d is associated with the Ii chain, which directs CD1d to the same compartment independently of the Tyr-based motif.209 The Ii chain needs to be degraded by cathepsin S to allow loading of the CD1d and to be directed to the cell membrane.210,211
of peripheral blood B cells and B cells of the marginal zone, and CD1d is expressed in intestinal epithelia and in a wide variety of hematopoietic cells. The members of the CD1 family are antigen-presenting molecules, and, after their synthesis, they must first reach an appropriate vesicular compartment for peptide loading on the way to the cell surface. There are striking differences in the intracellular distribution of the CD1 isoforms.201,202 CD1a is found mainly on the cell surface but also in early endosomes203,204 and in LCs that are present in the Birbeck granules. Together with CD1d, CD1b accumulates in the late endosomes and lysosomes, whereas CD1c is found in the early endosomes (transferrin receptor-positive). The ratio between surface expression and intracellular concentration also varies among the isoforms. The differential expression of the isoforms is regulated by a Tyr-based motif in the intracellular region that is present in all isoforms except CD1a.205,206 This sequence, YXXZ (Y, Tyr; X, any amino acid; and Z, an amino acid with bulky side chains), interacts with a member of the cytosolic adaptor proteins AP-1 through AP-4.207 AP-2 is positioned in the TGN and mediates delivery from the TGN to the endosomal-lysosomal system, whereas AP-2 is localized in the plasma membrane, where it directs proteins to clathrin-coated pits that lead to the entrance into the endocytic pathway. The AP-3 is implicated in the sorting of proteins from the TGN to endosomes and lysosomes. The AP proteins determine the specificity of destination for the CD1 molecules, but the binding to a specific AP member is determined by the residues that surround the Tyrbased motif.208 The YXXZ motif is essential for localization in the endosomal and lysosomal compartments, because mutations that are introduced into the sequence inhibit presentation of mycobacterial glycolipids to T cells.209 Although the YXXZ motif predicts the final destination, it does not indicate the route that is to be followed. The CD1d first follows a direct route from the ER to the cell surface, where, through the YXXØ motif (Ø, phenylalanine), it binds to AP-2. It is subsequently internalized and is directed to the MIIC compartment.209 MIIC overlaps considerably with lysosomes and may represent a fusion product between endosomes and lysosomes. CD1d is associated with the Ii chain, which directs CD1d to the same compartment independently of the Tyr-based motif.209 The Ii chain needs to be degraded by cathepsin S to allow loading of the CD1d and to be directed to the cell membrane.210,211
The antigens that are presented by CD1 molecules are lipids and glycolipids (Fig. 14.7). Mycolic acids are a class of long chain, branched, free fatty acids and constitute the predominant component of the thick outer layer of the mycobacterial cell wall. They confer the acid-fast staining property on mycobacteria. They contain approximately 80 carbon atoms and are known to be required for mycobacterial survival. In addition to mycolic acids, two glycolipids, lipoarabinomannan (LAM) and lipomannan, are large molecules that contain 20 to 100 glycosyl residues and are also presented by CD1 molecules.212 Other antigens include selected phosphatidylinositol mannosides and glycosylated mycolates, including glucose monomycolate.
Mycobacteria are taken up by phagocytosis and reside within nonacidified phagosomes (due to a paucity in the vacuolar H+ ATPase). Therefore, they are restricted in their capacity to fuse with late endosomes and lysosomes. The macrophage mannose receptor that has been implicated in glycolipid uptake by APC213 is absent from mycobacterial phagosomes.
The mechanism of distribution of various mycobacterial antigens to the subcellular compartments is not well understood. LAM has been found in late endosomes and lysosomes, either by being released from the phagosome or after uptake from the extracellular environment by the mannose receptor.213 Nevertheless, mycobacterial antigens are distributed throughout the endocytic system after their release from the phagosome.214,215 Antigens that are released from phagocytosed mycobacteria reach various compartments of the endocytic lysosomal system, where all CD1 isoforms, that is, CD1a, CD1b, and CD1c, gain access to mycobacterial glycolipids at these different sites (Fig. 14.8). CD1a and CD1c sample the antigens in phagosomes that are arrested in the early endosomal stage, whereas CD1b samples antigens in phagosomes that are arrested in the late mature phagolysosome stage. The CD1 molecule binds the lipid antigens by accommodating the first two lipid tails within the hydrophobic groove, whereas the hydrophilic caps (sugars) are exposed to the solvent and contact the TCR directly.216 The buried surface of the groove, as determined from crystallographic studies, is 1,400 Å, which could optimally accommodate a lipid with 32 carbons. However, some of the antigens that are presented by CD1 are 44 carbons long, and the mycolic acids are 80 carbons long. Presentation of large lipids takes hours (trafficking to late endosomes), whereas the same antigens with shorter chains are presented rapidly but inefficiently from the cell membrane.217 DCs specialize in the presentation of lipids with long alkyl chains, whereas nonprofessional APCs present preferentially short chain glycolipids. The acidic environment of the late endosomes and lysosomes is an important factor in the interaction of the large lipid antigens with the groove. The properties of the CD1b protein are dramatically altered in an acidic pH to facilitate the interaction with its ligands. There is a reversible unfolding of the α-helical portions of CD1 in an acidic pH, which permits the direct binding of the hydrophobic portions of the molecule, thus burying the alkyl chains.218,219 The depth, enclosed nature, and hydrophobicity of the interior of the CD1 groove is well adapted to carry out antigen presentation of lipids and amphipathic glycolipids, for which the hydrophobic component is sequestered within the groove, whereas the carbohydrates are left outside to interact with the TCR.
APC alters the structure of glycolipids by removing terminal glycosyl residues from certain glycolipids, resulting in the exposure of epitopes that are reactive with TCR.220 Glycosylation also generates new antigens, as is the case of glycosylation of mycolic acids during mycobacterial infections, with the use of glucose from the host and a mycobacterial enzyme.221 Glycolipid processing generates a large variety of lipid ligands for presentation by CD1 proteins.222
In addition to bacterial glycolipids (i.e., LAM) and glucose monomycolate, CD1b molecules recognize self-glycosphingolipids, such as GM1 ganglioside.223 Several of these gangliosides can bind to CD1b, and, in striking contrast to bacterial glycolipids, the self-glycolipids bind on the cell surface without the need of the late endosome and lysosome acidified environment.224 Five or more oligosaccharide groups are required to stimulate TCR recognition with a large component of the ligand that is exposed. Apparently, these gangliosides act as blockers to prevent harmful autoreactivity. The number of these autoreactive T cells increases in multiple sclerosis.223
CD1 System in Microbial Immunity
The protective value of CD1-mediated immunity is currently under intensive investigation.225,226,227 The CD1+ cells correlate with the level of immunity to Mycobacterium leprae and are present in the lesion tenfold more abundantly in the tuberculoid form (immunologically responsive) than in the lepromatous form (immunologically unresponsive). Low numbers of CD1+ cells correlate with low granulocyte-macrophage colony-stimulating factor (GM-CSF) levels and high IL10 levels, which inhibits GM-CSF secretion. GM-CSF is a key differentiating factor for mature CD83+ DCs.218,228 CD1-restricted T cells contribute to intracellular bacterial infections, such as M. tuberculosis, based on high levels of IFN-γ secretion and strong cytolytic activity.
The CD1 isoforms broadly survey the endocytic system over pathways that are distinct from those of MHC class I and MHC class II molecules, and, furthermore, they are restricted to presentation of an important class of antigens, lipids, that are not covered by the MHC molecules. Among them, CD1c is particularly
important, because it is expressed on LCs (in the absence of CD1b) and B cells (without CD1a or CD1b).229
important, because it is expressed on LCs (in the absence of CD1b) and B cells (without CD1a or CD1b).229
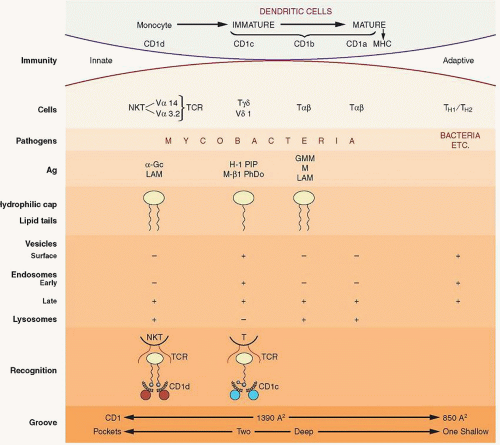 FIGURE 14.7. Microbial glycolipid presentation to T cells. The CD1 molecule, an orphan family of major histocompatibility complex (MHC)-like molecules, presents long carbon-chain glycolipids from mycobacteria and other similar pathogens, to natural killer T cells (NKT), T-cell receptor (TCR)-γδ, and TCR-αβ cells, linking innate and adaptive immunity. The CD1 molecules are expressed in various compartments of the endocytic system, providing a broad surveying mechanism for the detection of intracellular mycobacterial infections. Localization of CD1 molecules depends on endosomal targeting sequences for loading of glycolipids. The glycolipids in general consist of a hydrophilic head of carbohydrate molecules and one or two lipid tails. The head interacts with the TCR, whereas the lipid tails are accommodated within the large groove of the CD1 molecules, penetrating into two deep pockets. Ag, antigen; α-Gc, α-galactosylceramide; GMM, glucose monomycolate; H-1 PIP, hexosyl-1-phosphoisoprenoid; LAM, lipoarabinomannan; M-β1-PhDo, mannosyl-β1-phosphodolichols. |
 FIGURE 14.8. Migration of dendritic cells. A: A Langerhans cell, migrating through the labyrinthine space of the dermis, guided by chemokine signals that are received by its chemokine receptors. B: The Langerhans cell penetrates into the dermis through a hole (between the two groups of arrows) in the basement membrane (asterisk). (From Romani N, Ratzinger G, Pfaller K, et al. Migration of dendritic cells into lymphatics—the Langerhans cell example: routes, regulation and relevance. Int Rev Cytol 2001;207:237-270, with permission.) |
DENDRITIC CELLS
DCs, as their name indicates, are characterized by their long and elaborate cytoplasmic branching processes (the Greek word dendron means “tree”). The adaptive immune system, under evolutionary pressures, developed cells with exquisite specific receptors for sensing components of pathogens, so as to be able to generate molecular and cellular effector mechanisms for their elimination. Sensing of the pathogens or their products requires their breakdown (processing) and presentation by cells of the innate immunity. Foremost among the APCs are the DCs.230,231,232 DCs are a highly heterogeneous group that resides in most peripheral tissues at sites at which the body interfaces with the environment (i.e., skin, intestine, respiratory mucosa). Although DCs, as do other hematopoietic cells, ultimately derive from bone marrow progenitors, partially differentiated precursors are outside the bone marrow, such as peripheral blood, cord blood, and thymus. Such progenitors that are exposed in vivo to bacterial or inflammatory products, cytokines, and the like, differentiate to more mature cells with DC morphology and function. This large plasticity of DC development has created confusing and sometimes contradictory results. Basically, there are two lines of differentiation from stem cells, one along myeloid lineage and another along lymphoid lineage, thus the generation of two prevailing terms: myeloid DC and lymphoid DC. DC precursors circulate in the peripheral blood with a monocytic phenotype CD14+CD11c+CD13+. These cells, cultured in vivo in the presence of GM-CSF and IL4, give rise to the DCs that are considered as myeloid DCs. However, the same precursors cultured with fibroblasts differentiate to macrophages.233 IL6 that is released from fibroblasts in contact with the monocytic precursors up-regulates the macrophage colony-stimulating factor receptors. This switches differentiation to macrophages by an autocrine mechanism, based on the secreted macrophage colony-stimulating factor. Progenitors from cord blood that are considered lymphoid, with the potential to differentiate into NK cells, could give rise in vitro to phenotypically and functionally potent DCs under stimulation with various cytokines.234 In vitro studies may not represent the normal in vivo pathways of differentiation, but they nevertheless provide evidence that, in vivo also, partially differentiated cells of different lineages may choose a DC differentiation pathway when they are exposed to appropriate conditions. The stimuli for DC differentiation vary widely, from inflammatory microbial products235 to simply crossing endothelial barriers. DCs that develop within the thymus are CD8+ and are considered to be of lymphoid origin. However, it has been documented that even myeloid DCs can express CD8, so CD8 expression does not define lineage origin.236,237 Origin from lymphoid precursors was suggested by unique DCs known as plasmacytoid T cells or plasmacytoid monocytes. These cells have a typical plasma cell-like morphology; lack expression of myeloid markers; and are CD11c-, CXCR3+, and L-selectin (CD62L)+. DCs are heterogeneous with respect to their phenotype, anatomic distribution, and function.
Functionally, DCs have been distinguished into two groups, DC1 and DC2; the former derive from myeloid monocytes (pre-DC1), and the second derive from a plasmacytoid DC precursor (pre-DC2).238 This separation correlates with functional differentiation. Myeloid DCs from monocytes that are activated by the CD40 ligand (CD40L) produce large amounts of IL-12 and induce preferentially TH1 development. Lymphoid DC2s from plasmacytoid precursors produce lower amounts of IL12 and preferentially induce TH2 development.239,240 Another study, however, reached opposite conclusions,241 suggesting that the two DCs represent two separate evolutionary traits. The pre-DC1s express the Toll-like receptor (TLR)-2 and TLR4, and preDC2s express TLR7 and TLR9.242
Pre-DC1s are strongly positive for mannose receptor and rapidly produce large amounts of proinflammatory cytokines, such as TNF-α, IL6, and IL12, in response to ligands of TLR2-bacterial glycoproteins. Pre-DC2s produce IFN-γ in response to ligands of TLR9-bacterial DNA.243 The origin, response to stimuli, and pathways of differentiation of DCs vary greatly, depending on conditions that are still poorly understood.
The precursors of DCs from bone marrow progenitors move to the periphery as immature DCs and take position at crucial sites of potential entry of pathogens. Once they capture antigens and are exposed to stimuli, they mature and migrate to regional lymphoid organs to present antigens and to activate the adaptive immune responses by presenting antigens that are brought from the periphery. DCs are the professional APCs in immunity.
Migration of Dendritic Cells
DCs are cells that are continuously on the move from the moment they come out of the bone marrow to settle in various organs and tissues as immature DCs. In these locations, they are easily exposed to antigens and invading pathogens, and, now as mature activated DCs, they migrate to the lymphoid organs. Each of these pathways is orchestrated by distinct sets of molecules and receptors, foremost among them being the chemokines and their receptors.
Circulating monocytes and immature DCs express receptors for inflammatory chemokines (CXCR1, CCR1, CCR2, and CCR5), as well as receptors for bacterial and complement chemoattractants (receptor for N-formyl-methionyl-leucyl-phenylalanine-R and C5a-R),244 and, in response to such mediators, they develop into DCs.245 The crossing of the blood wall by DC precursors is mediated by a DC-specific adhesion molecule that is known as DC-specific intercellular adhesion molecule-3 (ICAM3) grabbing nonintegrin (DC-SIGN or CD209). DC-SIGN is a novel cell surface C-type lectin that is expressed on DCs, binds ICAM3, and mediates interactions between DCs and T cells.246 The extracellular region of DC-SIGN has a C-terminal, C-type carbohydrate recognition domain, particularly for mannose. DC-SIGN interacts with ICAM2 on the endothelial cells and establishes shear-resistant contacts with blood vessel walls for the transmigration of the DCs.247 The DC-SIGN-ICAM2 interaction is involved in the bloodborne DC precursor exit into peripheral tissues and in tissue mature DCs to peripheral lymphoid tissues. In the skin and epithelia of lung, intestine, and so on, immature DCs are exposed to stimuli that trigger their maturation, such as cytokines (TNF-α and IL1) and microbial products (e.g., lipopolysaccharide, LPS).
The mature DCs change to a new phenotype (MHC II3+, costimulatory and adhesion molecules positive) and acquire new migratory capacity. As a result, they migrate to the T-cell areas of the regional lymph nodes. DCs first down-regulate CCR1 and CCR5 (receptors for macrophage inflammatory protein [MIP]-1α and MIP-1β), up-regulate CCR7, and become responsive to secondary lymphoid tissue chemokine (SLC) and Epstein-Barr virus-induced molecule 1 ligand chemokine (ELC), the ligands for CCR7.248,249 Maturing DCs first move from their interstitial location to afferent lymphatic capillaries, where SLC is secreted from the endothelial cells and is carried to draining lymph nodes. The migratory pathways of the LCs, the DCs of the epidermis, provide a vivid example of the exquisite regulation of DC migration (Fig. 14.8).250
The LC precursors, CD11+ and CD1a+,200 are recruited into dermis, depending on the selectin expression of the dermal blood vessels.251 These cells express the CCR6 receptor, which binds the inflammatory chemokine MIP-3α (CCL-20), which is produced by keratinocytes of the epidermis.252,253 The LCs remain in the suprabasal layer, physically connected to neighboring keratinocytes by homotypic interactions that are mediated by E-cadherin, which is expressed by keratinocytes and LCs. Concomitantly, the CCR6 is down-regulated and the CCR7 is up-regulated, which
allows the LCs to move toward the lymphatic vessels in response to SLC. The basement membrane of the skin, however, stands as a barrier to their migration. LCs penetrate the lamina densa, which consists of type IV collagen and is digested by the matrix metalloproteinase of LCs. They cross the dermis and reach the abluminal side of the vessels. Entry into the lymphatic vessel is poorly understood, but P-glycoprotein may be involved.
allows the LCs to move toward the lymphatic vessels in response to SLC. The basement membrane of the skin, however, stands as a barrier to their migration. LCs penetrate the lamina densa, which consists of type IV collagen and is digested by the matrix metalloproteinase of LCs. They cross the dermis and reach the abluminal side of the vessels. Entry into the lymphatic vessel is poorly understood, but P-glycoprotein may be involved.
LCs are carried by the lymph and acquire morphology that is known as a veiled cell as a result of their broad sail-like membranous sheets. Entering the lymph node, they settle in the dependent areas in response to SLC (CCL21) and ELC (MIP-3β, CCL19), ligands for the CCR7 receptor. SLC is produced by the endothelial cells of the high endothelial venule, and ELC is produced by DCs or other cells in the paracortex.254 The LCs that settle in the paracortex are known as interdigitating dendritic cells (IDCs).
Accumulation of DCs around tumors follows the same mechanisms and depends fully on the interaction between chemokines released by tumors and the appropriate receptors on DCs 255.
Dendritic Cell Functions
DCs are best known for their efficient antigen presentation function. They capture antigens and pathogens, which they internalize and process for antigen presentation. DCs use a variety of receptors to take in antigens, such as receptors for Fc and complement, macrophage-scavenger receptors, mannose receptors, and DEC-205. However, of all the receptors that the DCs use to endocytose pathogens, the best studied is the DC-SIGN receptor that binds to HIV.256 HIV is not endocytosed by the DC, but the DC-SIGN binds to gp120 envelope glycoprotein, and the DC transfers the HIV to the lymphoid organs, where it enhances infection of T cells that express CD4 and chemokine coreceptors. DCs are efficient APCs for the class II-restricted pathway, but endocytosed antigens can also be diverted to the class I-restricted pathway. This is known as cross-priming or cross-presentation (see section “Presentation of Exogenous Antigen by Major Histocompatibility Complex Class I Molecules”). Immature DCs are crucial for this mechanism of presentation.
Interaction with pathogens by immature DCs not only prepares naive T cells for priming, it also activates immature DCs for maturation. This is mediated by signaling receptors, such as the Toll-like receptors of innate immunity (i.e., TLR2, which is involved in response to Gram-positive bacterial components, or TLR4, which interacts with LPS in response to Gram-negative bacteria). An interesting example of the diversity of responses to a single bacterial protein by immature DCs is the bacterial cell wall protein OmpA (from Klebsiella pneumoniae). This protein triggers maturation signaling of immature DCs through the TLR2 receptor, endocytosis via a receptor-dependent mechanism, and cross-priming of antigen-specific CD8+ T cells (in the absence of CD4+ T-cell help).257
DCs, however, are not simply antigen-delivering cells for lymphocytes. They play a key role in the maintenance of T and B lymphocyte pools in the absence of exogenous antigen. For T cells, this function depends on the high density of MHC class II molecules.258 It is postulated that the MHC class II molecules display self-peptides, and the interaction generates and transmits signals that are sufficient for the survival of T cells and could be traced from the cell surface to the nucleus (increase of IL12R β2 chain and IFN-γ mRNA). Contact between DCs and naive T cells in the absence of antigen is established by the interaction of DC-SIGN (DCs) and the ICAM3 (T cells).246 This strong interaction allows the establishment of interactions with other adhesion molecules to form the immunologic synapse. These synapses are antigen-independent and induce local Tyr phosphorylation, small Ca2+ responses, and long-term survival.259
DCs can regulate differentiation of naive B cells. In the initiation of the antibody response, a three-cell interaction takes place among T, B, and D cells with various combinations of cell-surface-expressed molecules. This gives time to B cells to process and present peptide through the class II MHC molecules, for a cognate T- and B-cell interaction.260 DCs directly modulate growth and differentiation of B cells and enhance IgG and IgA secretion by 30- to 300-fold, and through IL2 production stimulate IgM production. IL2 is a key cytokine that is produced early after DC stimulation by maturation signals.261
Dendritic Cells in Human Disease
DCs accumulate in the joints of rheumatoid arthritis patients and within psoriatic plaques, which express high levels of CD1b and CD1c, and are active stimulators of autologous T-cell proliferation. In contact allergy, hapten-modified proteins are processed by LCs that migrate to draining lymph nodes and initiate immune responses. A similar pathway is taken by DCs in the respiratory airways that capture and process inhaled antigens in asthma. The DCs are significantly higher in asthmatics. Targeting DCs may be an important new therapeutic approach for asthma.
In human parasitic infections, immature DCs can phagocytose the organism and restrain parasite replication in dermal infiltrates.
The role of DCs as potentiators and initiators of antiviral immune responses is well documented, especially in stimulating recall of cytotoxic T lymphocyte responses. Nevertheless, viruses still survive within DCs and subvert the immune response by down-regulation of MHC class I molecule expression, induction of immunosuppressive cytokines (IL10), or down-regulation of immunostimulatory cytokines (IL12). In the pathogenesis of HIV disease, the DCSIGN acts as a dendritic-specific HIV-1-binding protein that does not need a coreceptor and promotes binding and transmission of HIV-1 to T cells rather than entry to DCs. The virus is retained in an infectious state but is not allowed to enter the DC. DC-SIGN literally presents HIV to T cells in a nonprocessed infectious form. Various carcinomas display a heavy infiltrate of DCs with high MHC class II molecule expression that is associated with better prognosis. DCs have been used in trials in cancer therapy. Prospects for future successful DC immunotherapy are promising.262 T-cell immunity can be induced, even in patients with advanced stage IV melanoma, by vaccination with antigenpulsed mature monocyte-derived DCs.263 When the DCs are loaded with MHC class II molecule-binding melanoma peptides, strong tumor-specific TH1 responses were elicited.
Langerhans Cells
LCs were first observed in the epidermis by Langerhans and can be identified only by means of special stains that make use of the affinity of the cell for heavy-metal ions or the uptake by the cell of L-dopa, dopamine, and noradrenaline.266 The demonstration of a formalin-resistant and sulfhydryl-dependent ATPase is a reliable and specific technique for identifying LCs. Their ultrastructure was first described by Birbeck et al.,267 who also described a characteristic granule that bears Birbeck’s name.
LCs constitute 3% to 8% of all cells in the epidermis and are present in all parts of the skin, esophagus, and cervix; they are not found in the cornea, trachea, stomach, or bladder. In humans, the number of LCs per unit of skin varies between 40,000 and 100,000/cm2. Their precursors originate in the bone marrow, and in the mature stage they are located in the suprabasal layers of the skin, within the epidermis, where they can be distinguished from keratinocytes on the basis of absent desmosomes and tonofilaments and from melanocytes by the absence of melanosomes.
The long dendritic processes form a continuous network. The nucleus is irregular and lobulated. The cytoplasm is clear and contains microfilaments and multivesicular bodies, as well as the unique Birbeck granule, which is rod-shaped with periodic striations, giving it the appearance of a zipper. LCs are present in the dermal lymphatics, the marginal sinus, and the paracortex of draining lymph nodes, which suggests that these cells are mobile (see the previous discussion).
Mature LCs express a granule-associated antigen, E-cadherin,268 and HLA-D antigens. Ultraviolet irradiation depletes the skin of LCs and blocks the induction of contact sensitivity.
Veiled Cells
Veiled cells are present in the afferent lymph and resemble LCs.269 Their name derives from the possession of long, actively moving processes or veils that are approximately 100 nm thick. The veils do not contain organelles, and their movement must therefore be generated at a distance. Their nucleus is highly convoluted, and the cytoplasm contains bundles of microfilaments. Only approximately 4% of veiled cells contain a Birbeck granule, but all possess a large vacuole under the cell surface. They elicit a strongly positive response when they are tested for ATPase, and they resemble LC in the content of other enzymes. They are not actively phagocytic, but they possess Fc receptors.
After contact sensitization, Langerhans-like cells appear in the dermal lymphatics, and an increase in the number of veiled cells in the lymph that drain the area is noted. Once they are in the lymph node, most veiled cells localize in the T-cell-dependent areas, where they may function as interdigitating cells. Some veiled cells enhance the response of T lymphocytes to mitogens. Their function possibly consists of transporting antigens into the paracortical area, where they present them to T lymphocytes. Thus, a cellular chain for antigen transport exists in which more than one cell type takes part. Starting from the floor of the subcapsular sinus, antigen is localized first in cells that have the appearance of LCs, but, in deeper regions of the cortex, a second cell with morphologic characteristics of veiled cells has been identified.270 These two cells are similar to the cells that are known to migrate from skin areas to the paracortex.271
Interdigitating Dendritic Cells
The T-cell-dependent areas of the lymphoid organs contain substantial numbers of DCs that are known as IDCs.269 They have a clear cytoplasm and an irregularly shaped nucleus and extend long dendritic processes to contact T cells. They express MHC class II antigens but do not express receptors for the Fc fragment of Ig and complement. IDCs are positive for CD83 and the S100b protein (cytoplasm). The name usually is reserved to describe cells that lack Birbeck granules.
Many common features are shared by IDCs and DCs from other compartments (e.g., skin and splenic white pulp). This led to the view that a single DC system exists in which the peripheral nonlymphoid organs’ DCs migrate to the T-cell-dependent areas and become IDCs.272 This has been shown by labeling DCs that are isolated from the spleen of mice and injecting them into footpads. The labeled cells migrated to the periarteriolar sheath of the spleen or the paracortex of the lymph node.273 DCs from skin that is painted with hapten migrate within 24 hours to the lymph nodes.274
Similarly, Leishmania parasites that are injected into the skin are found within IDCs in the T-cell areas of the lymph nodes, probably as a result of uptake of the parasites by skin DCs that migrated to the lymph nodes to become IDCs.275 In patients with skin allergies, one finds Birbeck granules in some IDCs of local lymph nodes. Also, the T-cell areas stain strongly for CD12, a marker that is expressed on LCs. This evidence supports the notion that LCs migrate from areas of the skin to the T-cell areas of regional lymph nodes. Transportation of antigen from the periphery to lymph nodes is not only characteristic of the skin DC but is also a function that is performed by intestinal DCs and other DCs in the periphery.
Follicular Dendritic Cells
Follicular dendritic cells (FDCs) are found in the follicles and germinal centers (GCs) of lymph nodes and were identified by a distinct dendritic pattern of antigen localization in the follicles. Antigen trapping in the follicles requires antibody with an intact Fc fragment and complement. The antigen that is so trapped persists for long periods, in contrast to the antigen that is captured by the medullary macrophages. Cytologically, the FDCs have an irregular nucleus, a narrow rim of cytoplasm, and filiform dendrites, giving the cell a sunburstlike pattern. On their surface, they express Fc and C3 receptors and MHC class II antigens.
Electron microscopic studies have identified two FDC types: FDCs with filiform dendrites, which mature to FDCs with beaded dendrites.276 After secondary immunization, FDCs form spherical particles called iccosomes (immune complex-coated bodies; Fig. 14.9). The iccosomes are formed through interactions of the two FDC types. One cell binds immune complexes, and the second cell, with beaded dendrites, binds to the areas of the complexes. Thus, the bead becomes coated with the complexes and the cytoplasmic membrane of the first FDC that carries the complexes. At this stage, the bead is filled with immune complexes and is subsequently detached as iccosomes that are dispersed among the cells of a developing GC. This process constitutes what is called the alternative antigen presentation pathway,276 to distinguish it from the more conventional antigen capture and presentation.
Antigen complexes are poor immunogens because they deliver a down-regulatory signal to the B cell that is related to the phosphorylation of the immune receptor Tyr-based inhibitory motif on the B-cell Fc receptor. However, the FDC strongly expresses FcR, and it is conceivable that, as FDC binds to complexes, it limits the available Fc on the complexes for binding to the FcR on B cells. FDC also delivers co-stimulatory signals to B cells, probably through complement-derived fragments, C3dg and C3d, which are associated with the FDC-bound complexes. Complement fragments bind on B cells to complement receptor 2 (CR2, CD21), which is a member of the CD19/CD21/CD81 (TAPA1) complex, a coreceptor of the B-cell antigen receptor (see Chapter 12).277 Thus, the FDC-associated complexes colligate the antigen BCR and the CD19/CD21/CD81 coreceptor, providing two signals for B-cell stimulation.
The CD21 exists in two isoforms, one long and one short. FDCs selectively express the long isoform of CD21, whereas the short molecule is expressed by B cells. CD21 on FDC most likely participates in binding of complexes. The FDC-dependent B-cell stimulation is enhanced by IL2.
FDC traps and retains large numbers of retroviral particles,278 which markedly alter the function of FDC and the anatomy of the follicle. Infected FDC brings the virus and CD4+ T cells together, promoting their infectivity. The antigen presentation function of the FDCs is lost, and, eventually, the cells are destroyed.
There are two views on the origin of FDCs: one proposes a hematopoietic origin, whereas the other proposes a mesenchymalfibroblast origin. FDCs have a fibroblastic morphology and express certain molecules, such as vimentin and desmin, which are found in mesenchymal tissues. FDCs are also found in areas of chronic inflammation.279 As yet there is no convincing evidence for either theory. For the time being, FDCs continue to be defined as the cells in the follicles of lymphoid organs that have the property to trap complexes of Ig and antigen. The reticulum cells within the GCs were thought to be different from those in the mantle, but, by ultrastructural studies, DCs in GCs and those of the mantle zone (or primary follicles) are considered to be of the same family.280
 FIGURE 14.9. Follicular dendritic cell and iccosomes. A: Scanning electron micrograph that illustrates a follicular dendritic cell (arrow) with beaded dendrites. Each dendrite contains several iccosomes. B: Transmission electron micrograph of a lymphocyte from the germinal center with two iccosomes in contact with its surface (arrows). (From Szakal AK, Kosco MH, Tew JG. A novel in vivo follicular dendritic cell-dependent iccosome-mediated mechanism for delivery of antigen to antigen-processing cells. J Immunol 1988;140:341-353, with permission.) |
T-B-CELL INTERACTIONS
Immune responses are regulated by the dose and the localization of antigen.281 Furthermore, for T and B lymphocytes that need to collaborate, recognition of antigen by their antigen receptors is not sufficient to activate them, but a second signal (co-stimulation) is required. T lymphocytes are in the paracortical areas, and the most abundant APCs in the paracortex are the IDCs.
The complexity of the mechanisms that are involved in activation of naive T cells in vivo was elucidated with TCR transgenic T cells.282 When antigen is injected in the absence of an adjuvant, T-cell activation does not take place or is too small in intensity and short in duration and therefore is nonproductive. DCs, which are interspersed in the tissues behind the physical barriers of the body, are Nature’s adjuvants, and although they are immature, they are still phagocytic and able to capture pathogens or their products. Under these conditions and with stimulation by inflammatory cytokines, DCs transform into mature DCs, and although they may have lost their phagocytic capacity, they have acquired two new important properties. First, they change their chemokine receptors to migrate to the T-cell areas of the draining lymph nodes. In addition, they acquire co-stimulatory molecules (CD80 and CD86), adhesion molecules (CD48 and CD58), and class II MHC molecules that are loaded with peptides from newly internalized antigens expressed on the cell membrane. In addition to these molecular immunologic changes, morphologic signs of their activated state become apparent, with long cytoplasmic dendrites reaching out between T cells. This morphologic appearance contributed to their name as IDCs. Inflammatory cytokines, such as TNF-α, that are released from activated macrophages contribute to the activation of DCs283,284 and up-regulate CD40 expression on DCs,285 which further promotes interaction with CD40L+ T cells.286
Activated DCs lead to significant immune responses, whereas immature DCs can be tolerogenic, as has been shown with healthy human volunteers. Immature DCs inhibit CD8+ T-cell immunity to viral peptide-specific IL10-producing T cells.287 In contrast, mature DCs (triggered by a mixture of macrophage products, such as IL1β, IL6, TNF-α, and prostaglandin E2) induce functionally superior CD8+ T cells and polarize CD4 T cells toward IFN-γ production.
Maturational stimuli direct co-stimulatory molecules and MHC class II-peptide complexes to membrane microdomains (“lipid rafts”), where they are able to interact more efficiently with the CD28 and TCR as the DCs and T cells form the immunologic synapse (see Chapter 13).288,289 This is the first cognate interaction, which takes place between DCs and T cells in the paracortical area and results in full activation of T cells (Fig. 14.10). Co-stimulatory signaling amplifies the TCR signaling as much as 100-fold, as T cells respond to lower doses of antigen (˜100-fold lower dose). The co-stimulatory pathway up-regulates bcl-xL and prevents Fas-mediated apoptosis of newly activated T cells. Co-stimulation enhances production of IL2 from activated T cells and induces expansion of antigen-specific T cells.
The factors that drive these two processes are the levels of MHC class II-peptide complexes and, therefore, the TCR stimulation and the intensity of co-stimulation; both are related to the stability of the synapse.290,291,292 Recurrent and sustained exposures to antigen and to polarizing cytokines are essential for differentiation of CD4+ T cells.293 Early withdrawal of antigenic stimulation arrests differentiation, even in the continued presence of cytokines. For TH1 differentiation, prolonged signaling is required to induce appropriate epigenetic modifications to maintain high levels of T-bet expression (Chapter 13, see section “TH1 and TH2 Differentiation”).
An important aspect of T-cell differentiation involves antigen selection of a TCR-restricted repertoire. This was clearly shown in the immune response against pigeon cytochrome C, in which the specificity is determined by the CDR3 of the TCR α chain. Of eight preferred CDR3 features that are rapidly selected early during the response, only one (TCR-α93 glutamic acid) existed to any significant extent before immunization. This TCR restriction and clonal dominance is antigen-driven and is propagated by expansion of one or a few clones from the postthymic repertoire.
This selective T-cell expansion for effector and memory function takes place in the T-cell zone, before the GC reaction. Inflammatory cytokines, particularly IL1 or TNF-α, also mediate some of these effects through CD28 co-stimulation by up-regulating expression of CD80 and CD86 on DCs.
After the peak of clonal expansion, the number of antigenspecific T cells falls owing to cell death, which may occur via Fas-dependent or activation-induced cell death. For the antibody response to continue, T and B cells, which are specific for different
epitopes of the same antigen, must move from their separate locations to meet. Of the surviving activated T cells, most of them move out of the lymph node as memory T cells, but a small portion migrates to the edges of the paracortex next to the follicle, and B cells also move to the same location.294,295
epitopes of the same antigen, must move from their separate locations to meet. Of the surviving activated T cells, most of them move out of the lymph node as memory T cells, but a small portion migrates to the edges of the paracortex next to the follicle, and B cells also move to the same location.294,295
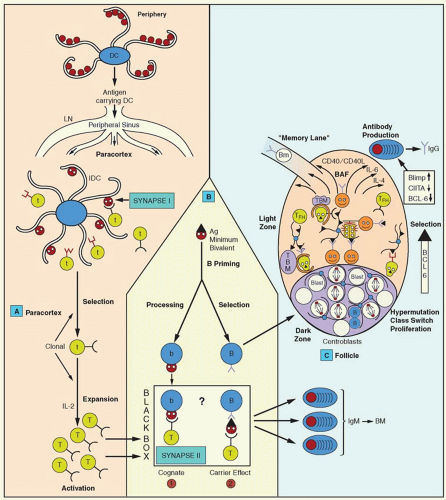 FIGURE 14.10. Chance and necessity: the germinal center reaction. Antigen (Ag) captured in the periphery by dendritic cells (DCs) is brought to the regional lymph nodes to be presented to naive T cells (t) (synapse I). Clonally selected naive T cells become activated by interleukin (IL)-2 (T). They up-regulate the CXCR5 receptor, respond to BLC chemokine that is secreted from follicular cells, and move toward or even inside the follicle. In the border between the paracortex and the mantle of the follicle, they interact with B cells. The interactions are not yet well characterized in vivo. Some B cells process antigen and may present it to T cells that are selected by DCs (synapse II) (1). B cells may have been selected by the same antigen but for a different epitope. B and T cells may bind to the same immunogen but on different epitopes (“carrier effect”; synapse II) (2). Synapse II promotes differentiation of plasma cells that secrete immunoglobulin (Ig) M. B cells move inside the follicle to the pole that is adjacent to the T-cell zone (dark zone) and begin intensive proliferation. They are Ig-negative and are called centroblasts. B cells undergo mutations in the V gene at a high rate (somatic hypermutation) and change their IgM to another Ig class (class switch). When proliferation subsides, centroblasts move to the center of the germinal center—i.e., the light zone—and are known as centrocytes (Ig-positive). Their B-cell receptor (BCR) is submitted to the scrutiny of antigen-antibody complexes on dendrites of DCs of the densely populated light zone. In most centrocytes, somatic hypermutations have decreased or destroyed the affinity of their BCR and enter the pathway for apoptosis. Their corpses are removed by macrophages known as tingible body macrophages (TBM). If the affinity is increased, they are given the green light for survival and growth (BAFF, IL-6, IL-4, CD40-CD40L). Surviving cells have higher affinity than their predecessors (affinity maturation). They receive signals from germinal center DCs or follicular T-helper cells (TFH). Depending on the instructions that are received, some centrocytes go down memory lane, whereas others become plasma cells. Two genes are most influential in the regulation of germinal center reaction: B-cell lymphoma 6 (BCL-6) in the early phase represses all genes that regulate the second state; the B-lymphocyte-induced maturation protein-1 (Blimp-1) is released from repression when BCL-6 is down-regulated by high-affinity BCR signals. Blimp-1 regulates plasma cell differentiation. b, naive B cell; B, primed, antigen-specific B cell; BAF, B-cell activating factor of the TNF family; BM, bone marrow; CIITA, major histocompatibility complex class II trans-activator molecule; IDC, interdigitating dendritic cell. |
Chemokines and their receptors are the main regulators of primary immune responses, setting the stage for the main players as well as orchestrating their performance. CCR7 is the most important for organizing the appropriate microenvironment to make the initial interaction possible.296 As multiple cells contribute to an immune response, change in the chemokine receptor program regulates the second stage in the cell interactions for antibody synthesis. CCR7 is the receptor for chemokines SLC (6Ckine, exodus2, and CCL21) and ELC (MIP-3β, exodus3, and CCL19). SLC is secreted by the cells of high endothelial venules and mediates the crossing of T cells to the lymph nodes. ELC is secreted by stromal cells of the paracortex and attracts naive T cells to settle within the T-cell areas (see Chapter 11). DCs in the peripheral tissues up-regulate CCR7 expression as part of their maturation and are guided to the paracortical areas. The CXCR5 is the receptor for chemokine BLC (CXCL13), which is secreted by stromal cells in the follicles and attracts B cells that cross the high endothelial venules to settle in the follicular area. Activated CD4+ T cells up-regulate CXCR5 and migrate to the follicles.297,298,299,300,301 Whether the T-cell-B-cell interaction takes place on the paracortical side295 or the follicular side294 of the T-cell-B-cell border is not clear. The fact is that the two cells physically interact (second cognate interaction), forming a synapse by adhesion molecules and supported by co-stimulatory molecules, including the inducible costimulator (ICOS) on activated T cells and its counterreceptor B7-h on B cells.302
GERMINAL CENTER REACTION
Interaction of B cells with activated T cells initiates their proliferation and migration into the area within the primary follicle, which is now called the secondary follicle. Some of the activated B cells differentiate at the edge of the T-cell zone into plasma cells, secreting IgM and migrating to the medullary cords and on to the bone marrow. Plasma cell differentiation in this area could also be induced in a T-cell-independent way, especially from antigens known as T-cell-independent antigens. The IgM antibody is part of the early phase of antibody production.303 The remaining antigenspecific B cells are recruited within the follicle and rapidly expand to form the germinal centers.304 GCs are formed as a result of the expansion of one or a small number of B cells and are therefore monoclonal or oligoclonal. The expanding population of B cells fills the FDC network,305 and although T cells facilitate their formation, GCs may be formed in the absence of T-cell signals for certain antigens.306 Multiple steps of mutation and selection are the mean driving forces in the formation of the GCs, and recirculation of positively selected centrocytes is quite unlikely.307 CD27 is important for GC formation by two distinct mechanisms. T cells support formation of GC as a result of interaction of CD28 (T cells) with CD27 (B cells) (centroblast expansion) and, in addition, CD27+ T cells may be interacting with CD70 on B cells.308 The GCs are oligoclonal and with a cell cycle time of the dividing B cells of approximately 6 hours, the number of accumulating blasts within 3 days is of the order of 1 to 1.5 × 104. During this exponential growth, the small recirculating B cells are excluded and are pushed aside to form the familiar mantle that surrounds the GC. By the time the B blasts finish their exponential growth, they have filled the FDC network that is close to the T-cell zone, which is known as the dark zone, and the B blasts are known as centroblasts. Centroblasts are Ig– and give rise to nondividing Ig+ cells, which are known as centrocytes, which occupy the center of the follicle or the light zone. The light zone has been divided into an apical (FDC CD232+) and a basal component (FDC CD23–; Fig. 14.11).
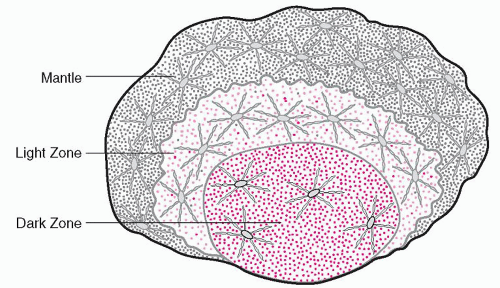 FIGURE 14.11. The germinal center. Dark zone, centroblasts in cell cycle. Light zone, strong expression of CD23 on dense follicular dendritic cell (FDC) network. Mantle, loose FDC network. |
Several important events for the life of B cells take place in the GCs: clonal expansion, somatic hypermutation (SHM) resulting in affinity maturation, Ig class switch, and generation of memory B cells and plasma cells. All naive B cells express the CXCR5 receptor for chemokine CXCL13 (BCA-1, B-cell attractant [chemokine 1]), which is secreted constitutively by cells within the DC network in the follicular area. Furthermore, the DC-specific chemokine CD-CK1 (CCL18, also known as PARC) preferably attracts CD38– mantle-zone B cells toward the FDC and may well provide the initiation signal for the formation of GCs.309 Responsiveness to the CXCL13 is also shown by a minor subpopulation of memory CD4+ T cells that express the CXCR5 receptor for the CXCL13 chemokine and, as a result, are attracted to the follicles; they are known as follicular T-helper cells (TFH).297,298 The expression of CXCR5 is transient and occurs during T-cell activation but before their proliferation. The CXCR5+ T cells constitute only a minor proportion of peripheral T cells. They also express the CC-chemokine receptor 7 (CCR7) and L-selectin (CD62L), allowing a steady-state re-circulation through secondary lymphoid organs.299 Several molecules expressed on TFH cells mediate help to B cells, but crucial among them are CD40L (CD40 ligand or CD154) and ICOS (inducible T-cell co-stimulator), which interact with CD40 and the ICOS-ligand (ICOS-L), respectively, on B cells. Another important molecule of TFH is SAP (its name derives from its association with an activation molecule known as SLAM [CD150], or signaling lymphocytic activation molecule). A significant member of TFH undergoes apoptosis, as shown by gene expression profiling.300
The FDCs are strongly positive for CD21 and CD23 and form a dense network in the light zone. In the dark zone, they are weakly positive for CD21 and negative for CD23, and they form a light network. The FDCs express the long isoform of CD21, which is the only specific marker for human FDCs, whereas the B cells express only the short isoform. FDCs trap immune complexes and present antigen to B cells, although the B cells can undergo somatic mutation and memory cell formation, even in the absence of complexes,310 probably by other signals that are received from FDCs, which support B cells in the immune response in several ways. Engagement of CD21 in the BCR by complement in the antigen-antibody complexes augments the stimulation that is delivered by FDCs.311 Furthermore, engagement of the FcγRIIB on DCs diverts binding of the complexes from the same receptor on B cells. FcγRIIB is an inhibitory receptor, and, thus, by trapping the complexes, the FDCs protect B cells from inhibition.311
There is cross-talk between FDC progenitors and B cells, because B-cell signals contribute to FDC maturation and network development. LT-α, LT-β, and TNF-α from B cells, and TNFR-1
expression by some nonbone-marrow-derived cells lead to FDC cluster formation in the lymphoid organs.312
expression by some nonbone-marrow-derived cells lead to FDC cluster formation in the lymphoid organs.312
Because FDCs have no phagocytic activity and do not synthesize MHC class II molecules, they are unable to present peptides that are complexed with the MHC after processing of internalized antigens. However, there are two other ways that FDCs perform their antigen-presenting function. Cytochemically, antigen has been detected on FDCs along filiform or beaded dendrites. The beads, like pearls on a string, are particles that contain the immune complexes called iccosomes (immune complex-coated body), which are usually detected in the early phase of the formation of the GCs.276 Complement in the complexes binds to CD21 on FDC and delivers a crucial signal that dramatically augments the B-cell stimulation resulting from binding of the antigen to BCR. FDCs have no phagocytic activity and do not synthesize MHC class II molecules, and therefore they are unable to process antigen for presentation.313 MHC class II, however, has been detected on the surface of FDCs in the form of microvesicles that contain MHC class II molecules and other antigens that are foreign to the FDC.314 These vesicles are exosomes that are enriched in MHC class II molecules and members of the tetraspan family (i.e., CD37, CD53, CD63, CD81, and CD82; see Appendix A). The exosomes derive from multivesicular endosomes that are released by B cells with specificity for attachment to DCs and the ability to stimulate CD4+ T cells.315,316 This is an interesting example of cell communication, antigen processing, and presentation for T-cell activation. FDCs therefore have a dual mission for B-cell response within the GCs: antigen processing and presentation to B cells, and promotion of B-cell differentiation and isotype switching.317,318
In addition to FDCs, T cells have a critical role in GC formation, primarily for the late stages of GC reaction and not during the initial development.319,320 GCs that are induced in the absence of T cells are of short duration, and the V genes of the antibodies do not undergo hypermutation. The GCs abort dramatically at the time when T cells normally select the high-affinity B cells.319 The demise of the GCs at this point is a fail-safe mechanism to prevent autoreactive B cells from escaping into the periphery, with development of autoimmunity.
In the dark zone, lymphocytes are densely packed, and only fine FCD processes penetrate this area. The FDCs in the dark zone are CD23– and only weakly CD21+. The centroblasts are in rapid cell cycle and do not increase in number, but they give rise to a progeny of centrocytes that are nondividing and express surface Ig. Centrocytes are located in the light zone and are CD21 strongly positive and CD23 moderately to strongly positive. In this stage of the evolution of the GC, the B cell is selected for survival or death, based on the affinity of the antigen receptor as the cell emerges from changes induced by hypermutation (see the following discussion).
B Cells in Germinal Centers: Struggle for Survival
Proliferation of B cells in the GCs is stimulated by antigen, but different mechanisms regulate the proliferation in the two GC zones, dark versus light. The paucity of T cells in the dark zone suggests that this early, expansive B-cell phase is supported by strong stimulatory signals from complement-containing immune complexes, FDC-B-cell cognate interactions, and soluble factors from FDCs.321 In the light zone, TFH cells play a more important role and, although the density of FDCs is much higher, some FDCs are inhibitory for B-cell activation. GCs develop within 4 to 6 days after primary immunization, and centroblasts in the dark zone continue to maintain a high output of centrocytes for 7 to 14 days and undergo SHM that results in antigen receptor diversification.
The decision between life or death, a process known as selection, takes place within the light zone as the B cell emerges from the dark zone. The mechanism of selection is not yet clear, but antigen that is trapped on FDCs is used to “test” the affinity of the BCR of the centrocytes. A key determinant for selection is the CDR3, which has a major contribution to diversity and affinity of the receptor. Structurally identical receptors are handled differently if variations in CDR3 exist between them, and such variations drive the selection process.322 The vast majority of these random mutations is deleterious to antigen binding and destroys or diminishes the affinity of the BCR that existed when the antigen-specific B blast entered the dark zone from the paracortex-follicular border. These cells are negatively selected and are diverted to apoptosis.
Other cells have acquired autoreactivity, and these cells receive no signals from FDCs and therefore die from “neglect.”323 Of the cells in which BCR expresses reactivity with antigen, some emerge from the mutational process with high affinity, and only those are positively selected to achieve affinity maturation. Affinity maturation was discovered 42 years ago324 but remained poorly understood until somatic diversification of BCR genes in the GCs was demonstrated.325 Affinity maturation signifies the increase of the affinity of antibodies between those that are produced early after primary immunization (low affinity) and those that are produced 2 weeks after immunization or, better, after secondary immunization (high affinity). Affinity maturation is associated with somatic diversification that occurs during hypermutation. As shown by studies in mice, the D and J regions of the H chain (16 amino acids long) of the antibodies that are synthesized 7 days after immunization have no residue changes that could be attributed to mutations. However, by 14 days the antibodies carry somatic mutations that have resulted in increase of the affinity.326 Positive selection of these high-affinity antibodies constitutes affinity maturation.
Survival of the Fittest
One of the puzzling phenomena related to the life of B cells after their release from the bone marrow is the fact that approximately 70% of them do not reach maturation and die. B-cell survival and maturation relies on the delivery of signals, most importantly through BCR, and survival depends on a mechanism of positive selection.327 The immature B cells that leave the bone marrow undergo a progressive maturation process that takes the cell through three stages, known as transitional stage type 1 (T1), type 2 (T2), and type 3 (T3).328 T1 cells are IgMhi, IgD–, CD23–, and CD21lo, and T2 cells are IgMhi, IgDhi, CD23+, and CD21hi.329
Although the location and mechanism by which this huge number of B cells perish have not been identified, it is possible that a prime location is the light zone, in which the mechanism of cell death is by apoptosis as a result of a BCR that is incapacitated by hypermutation.
B cells require and receive multiple signals from different cells and molecules for survival. The cognate interaction between FDCs and B cells provides signals through antigen presentation (albeit weak), as well as through several receptor-ligand pair interactions, such as ICAM-lymphocyte function-associated antigen-1, vascular cell adhesion molecule-1-very late antigen-4, and CD23-CD21.330
In addition, DC stimulates B-cell proliferation and Ig synthesis, which are mediated by the CD137-ligand system.331 CD137 is a member of the TNF receptor family that is expressed by FDCs and stimulates the CD137 ligand-expressing B cells by reverse signaling.
Germinal Center B Cell
An important B-cell survival factor that is known as the B-cell activation factor member of the TNF family (BAFF or TNFSF13B) promotes maturation of B cells and prolongs their survival and, under certain conditions, B-cell growth.332,333 BAFF forms a trimerlike TNF, like other members of the family, but its unique
feature is the presence of two Mg2+ ions. The gene for TNFSF13B (BAFF) is located in human chromosome 13q32-34, a location that is frequently involved in translocations in Burkitt lymphoma. BAFF exists as a membrane-bound form and as a soluble form. It is closely related (approximately 50% similarity) to another TNF family member, a proliferation-inducing ligand (APRIL). Both BAFF and APRIL bind to two receptors, B-cell maturation antigen (TNFRSF17) and transmembrane activator, and calcium modulator and cyclophilin ligand interactor (TNFRSR13B or TACI), with high affinity, but preferences by the ligands have been detected,334 with BAFF being associated more often with TACI than with TNFRSF17.335 A third receptor, called BAFFR (TNFSF13C), selectively binds BAFF and not APRIL. However, a basic amino acid sequence close to the NH2 terminus of APRIL binds to proteoglycans in the extracellular matrix or cells and produces its oligomerization. It is probably an APRIL-specific receptor.336 TNFSF17 (previously known as BCMA), TNFRSF13B, and TNFSF13C (BAFFR) are located on human chromosomes 16p13.1, 17p.11.2, and 22q13.1-13.3, respectively.
feature is the presence of two Mg2+ ions. The gene for TNFSF13B (BAFF) is located in human chromosome 13q32-34, a location that is frequently involved in translocations in Burkitt lymphoma. BAFF exists as a membrane-bound form and as a soluble form. It is closely related (approximately 50% similarity) to another TNF family member, a proliferation-inducing ligand (APRIL). Both BAFF and APRIL bind to two receptors, B-cell maturation antigen (TNFRSF17) and transmembrane activator, and calcium modulator and cyclophilin ligand interactor (TNFRSR13B or TACI), with high affinity, but preferences by the ligands have been detected,334 with BAFF being associated more often with TACI than with TNFRSF17.335 A third receptor, called BAFFR (TNFSF13C), selectively binds BAFF and not APRIL. However, a basic amino acid sequence close to the NH2 terminus of APRIL binds to proteoglycans in the extracellular matrix or cells and produces its oligomerization. It is probably an APRIL-specific receptor.336 TNFSF17 (previously known as BCMA), TNFRSF13B, and TNFSF13C (BAFFR) are located on human chromosomes 16p13.1, 17p.11.2, and 22q13.1-13.3, respectively.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


