Pituitary Development
ORIGIN
Phylogenetic studies in several vertebrate species led to the conclusion that the pituitary gland arises from oral epithelia. Fate-mapping experiments conducted in these animal species trace the origins of the pituitary gland back to the neural plate. In studies of grafting quail chick chimeras, the origin of the pituitary was localized to the midline of the anterior neural ridge. By means of surgical ablation performed in chick embryos, the rostral ridge of the neural plate was identified as the source of cells that give rise to pituitary tissue.4–6 In amphibians, tracing experiments have confirmed the neural origin of pituitary gland7,8 and similar conclusions have been reached about zebrafish.9,10 Additionally, by focalized application of a carbocyanin dye, DiI, into the rostral end of the neural plate at the open neurula stage (9.5 days postcoitus) in rats, labeled cells could be identified in Rathke’s pouch, and they could develop into the secretory cells of the adenohypophysis in 7 additional days.11 Thus evidence indicates that the anterior neural ridge is the origin of Rathke’s pouch, which eventually gives rise to cells of the pituitary gland. Subsequent to the folding of the embryonic head, the anterior neural ridge is displaced ventrally to form the portion of the oral epithelium that later gives rise to the roof of the mouth and additional structures, including the pituitary gland. Consistent findings in many species make it apparent that the process of pituitary development is, for the most part, evolutionarily conserved from lower vertebrates to higher mammals.
ONTOGENY
In humans, the anterior lobe of the pituitary gland originates from an invagination of the stomodeal epithelium termed Rathke’s pouch.12 The stomodeal epithelium that contains the pituitary primordium is formed by the third fetal week, and the invagination of stomodeal epithelium occurs dorsally to form Rathke’s pouch by the fourth week. The formation of Rathke’s pouch is complete and disconnected from the oral epithelium by the end of the sixth week of fetal life.13 In parallel, the hypothalamus is the first region of the forebrain to differentiate. From 4 weeks, the hypothalamic sulcus, chiasmatic plate, and mammillary bodies are recognizable. These two organs, hypothalamus and pituitary, develop interdependently.14
Similar to the ontogeny observed in humans, Rathke’s pouch in mice is derived from an anlage that arises as an upgrowth from the lining of the oral cavity’s roof. At its earliest stage, the murine pituitary primordium is defined as an intimate point of contact between the neural ectoderm and the oral roof ectoderm on embryonic day 8.5 postcoitus (e8.5), which marks the first event in the pituitary’s development. Organogenesis of the adenohypophysis begins as the cells of the pituitary placode in the oral ectoderm thicken and invaginate to form the nascent pituitary. In the e9.5 mouse embryo, this anlage can be seen located rostrally to the oropharyngeal membrane. Dorsal movement of the epithelial layer from the roof of the mouth induces a cone-shaped intrusion dorsally as Rathke’s pouch, or the adenohypophyseal pouch. Before the formation, a developmentally important molecular marker, Sonic hedgehog (Shh), is expressed uniformly in the oral epithelial layer. The expression of Shh is excluded before the intrusion of pituitary anlagen can occur in the e9 mouse embryo.15 Rathke’s pouch thickens as development proceeds and elongates dorsally relative to the oral cavity by the stomodia-adenohypophyseal channel. By e10.5 in the mouse, Rathke’s pouch has formed as a rudimentary structure and separated from the ventral pharyngeal epithelium.
At the time Rathke’s pouch is pinched off at e11 in mice, the first round of accelerated mitotic activity is initiated in the anlagen.16,17 In the ensuing patterning period, mitotic activity is observed most prominently in the rostral part of Rathke’s pouch, with several buds emerging and enveloping areas of vascularized mesenchyme. Progenitors of the hormone-secreting cell types arise from the ventral proliferation of cells, and this region of rostral Rathke’s pouch eventually gives rise to the anterior lobe, or the pars distalis. The dorsal aspects of Rathke’s pouch, in contact with the descending infundibulum processes and rostroventrally with the hypophyseal cleft, remain thin and form the intermediate lobe, or the pars intermedia. Anterior pituitary cell types are positionally determined as they initially emerge from proliferation zones,15,18 with the somatotrope/lactotrope cells arising caudomedially, gonadotrope cells more rostroventrally, corticotrope cells ventrally, and melanotrope cells dorsally. This pattern of pituitary development is generally similar in most mammals.
CELL LINEAGE DETERMINATION
Endocrine pituitary cell types in the adenohypophysis are derived from a single population of cells. The initial expression of pituitary hormone genes marking the terminal differentiation events of individual cell types occurs in a sequential manner. In mice, POMC gene expression emerges as the first pituitary marker at e11.5 and can be detected in the anterior pituitary by e13.5. However, the fate of cells that will give rise to those five different anterior pituitary cell types is determined prior to the initial pituitary POMC expression. In tissue-culture experiments where pituitary anlagen were taken and placed in a culture away from the influence of the diencephalons, pituitary anlagen taken at e11 were capable of generating cells expressing all five anterior pituitary hormone genes, while anlagen taken at e9.5 required additional growth factors, with the exception of corticotrope, which always differentiates regardless of the culture medium.19 Critical events occur at the time pituitary anlagen become committed to developing into pituitary precursors that will subsequently express pituitary genes that become regulated in a cell-autonomous fashion.20 The timing of this commitment event is coincidental with the formation of Rathke’s pouch.
As an anlage, Rathke’s pouch is the source of all endocrine pituitary cell types. In mice, after the initial appearance of corticotrope, expression of GH gene can be detected by e15.5, followed by thyrotropins, gonadotropins, and PRL. Gene expressions of all anterior pituitary hormones are detectable by e17.5, with the exception of PRL, which can be consistently seen by the time of birth (e19 in the mouse). Another early marker of Rathke’s pouch is αGSU, and the transcripts are detected throughout Rathke’s pouch by e9,21 although they are confined to the rostral tip of the anterior lobe by e12.5 and ultimately restricted to thyrotrope and gonadotrope from late gestation through adulthood. Following proliferation and early organ expansion, a series of different cell types arise in a distinct spatial and temporal fashion. Table 8-1 provides a time line of the initial expression of pituitary hormone genes in several species.
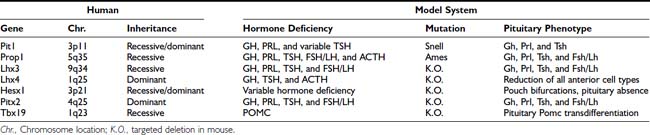
Transcription Factors and Pituitary Development
Parallel to the sequential emergence of pituitary cell types, a series of homeodomain family transcription factors are expressed as the adenohypophysis is becoming committed. With improved molecular genetic techniques, functional studies of these transcription factors in animal models, particularly in mouse models, have established molecular mechanisms underlying development of the pituitary gland. The expression profiles of Hesx1, Lhx3, Lhx4, Pitx1/2, Prop1, and Pit1 homeodomain factors, in addition to the expressions of Tbx19 and GATA2, dictate the commitment, determination, and differentiation events of the pituitary gland. These genes were initially studied in animal model systems that arose either from naturally occurring mutations or were created by reverse genetic techniques. Without exception, phenotypes observed in each animal model system are also observed in human cases with defects in the corresponding orthologous genes (Table 8-2). The phenotypes observed in human cases range from single pituitary hormone deficiency to combined pituitary hormone deficiency (CPHD) affecting several pituitary hormones in addition to GH. Study of the development of the pituitary gland serves as a model of progressive restriction in gene expression, and the pituitary gland has become a prototypic model organ system to study organogenesis, cell type determination, and differentiation.
PIT1 GENE
The Pit1 gene (POU domain, class 1, transcription factor 1 [POU1F1]) encodes a 33-kD, 291-amino acid transcriptional activator that is capable of DNA binding and transactivation, and it was initially isolated by its ability to bind to the responsive element of the GH gene promoter.22,23 Pit1 is expressed exclusively in the pituitary gland. In mice, the initial expression of the Pit1 gene transcripts can be detected by e13.5, exclusively in the anterior ventral pituitary (Fig. 8-1). The expression of Pit1 persists in adults and co-localizes with expression of GH, PRL, and TSHβ genes. Further studies revealed that the product of the Pit1 is capable of binding to responsive elements in the promoters of the GH gene,24 the growth hormone–releasing hormone receptor (GHRHR) gene,25 the PRL gene,26 and the TSHβ gene. The Pit1 protein is also capable of binding to the responsive elements of the Pit1 gene itself and is required for the continued transcription of the Pit1 gene.27 The structure of the Pit1 gene is evolutionarily conserved and is found in mouse, human, and all other vertebrate animals examined, although Pit1 may play diverse functional roles in different physiologic pathways in individual species.
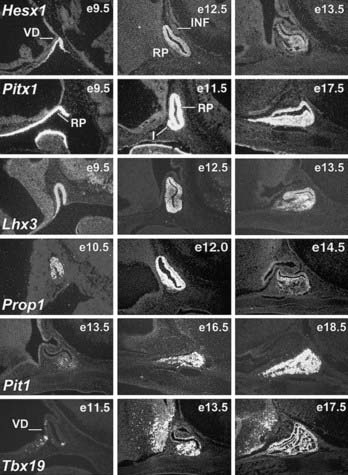
FIGURE 8-1. Expression of selected transcription factors in pituitary development by in-situ hybridization. Expression of Hesx1, Pitx1 and Lhx3 are detected in Rathke’s pouch (RP) at mouse embryonic stage e9.5 and are maintained at e12.5, after which Hesx1 expression is rapidly extinguished while Pitx1 and Lhx3 continue to be expressed. Prop1 expression initiates at e10.5, reaches maximum intensity at e12.5, and attenuates at e14.5. Pit1 expression initiates at e13.5 and is maintained throughout pituitary development and adulthood. Initial Tbx19 expression can be observed in the ventral Rathke’s pouch and ventral diencephalon (VD) at e11.5, and its expression is maintained.
Animal Model
Snell mice28 are a well-studied animal model of pituitary function, which arises from a spontaneous single nucleotide mutation in the Pit1 gene that results in the substitution of W261C in the homeodomain, rendering the mutant gene product incapable of DNA binding and hence unable to activate potential target genes.29 Mice heterozygous for this mutation are phenotypically normal. The homozygous offspring of this mutation are dwarf and infertile, and they exhibit loss of three pituitary hormone cell types, GH, PRL, and TSHβ; whereas the gonadotrope and corticotrope cells are unaffected, suggesting that the Pit1 is required for terminal differentiation of the somatotrope, lactotrope, and thyrotrope cell types. In the Pit1Snell animal model where the Pit1 gene is functionally defective, the initial activation of the Pit1 is unaffected, while the later transcription of the Pit1 gene is altered, resulting in the failed expression of the Pit1 in the adult animal and a dwarf phenotype.29,30 The Pit1 lineage can be converted to alternative fates before e17.5 but exhibits a cell-autonomous commitment after e17.5, when Pit1 gene regulation shifts from a Pit1-independent early enhancer to a Pit1-autoregulated later enhancer.31 Pit1Jackson is a second mouse model with a defect in the Pit1 gene. The genomic structure of the Pit1 gene, located on chromosome 16, is grossly rearranged in mutant Pit1Jackson mice, with a phenotype very similar or identical to that of the Pit1Snell mice.29 In addition, Pit1 mutations result in decreased activity of the insulin/IGF1 pathway, which may result in physiologic homeostasis consequences that favor longevity32,33 (for reviews see [34–36]).
Related Diseases
The human Pit1 gene has been mapped to chromosome 3. Lesions in Pit1 have been identified as an etiology of CPHD (see Table 8-2). Initial study has revealed a homozygous nonsense mutation R172X in the Pit1 gene in a patient of consanguineous parents with cretinism due to deficiency of GH, PRL, and TSHβ.37 Many cases of CPHD with Pit1 defects have since been reported. It appears that the inheritance of Pit1 mutations in humans is complex, ranging from autosomal recessive to autosomal dominant to imprinting with variable phenotypic penetrance.38 Pituitary gonadotropins and corticotropins are normal in Pit1-defective patients. Deficiency of GH is consistently observed in all Pit1 patients, and deficiency for PRL is observed in most patients, whereas TSHβ deficiency usually has a delayed onset and incomplete penetrance (see Table 8-2). Different backgrounds may be the major contributing factor to the TSH phenotypic variation. Alternatively, however, there exists an embryonic population of thyrotrope termed rostral tip thyrotrope. The expression of this embryonic TSH is not Pit1 dependent, and consequently it may be a contributing element to the TSH phenotypic variation observed in Pit1 patients. The presentation of patients with Pit1 disorders varies considerably. At infancy, they usually have a protruding forehead, depressed facial structures, and a saddled nose, although CPHD is generally not diagnosed until growth retardation due to the deficiencies of GH and thyroid hormone becomes obvious.39,40
Mechanism
The modular structure of the Pit1 protein can be divided into the transactivation and the DNA-binding domains. The transcriptional activation domain is located in the first 80 amino acids, followed by a POU DNA-binding domain at the C terminus. The POU domain is further divided into a 75-amino acid POU-specific domain, which is conserved among various POU-domain proteins, and a 60-amino acid POU homeodomain with a linker region between them. The POU homeodomain by itself is sufficient for low-affinity DNA binding, although both the POU-specific domain and POU homeodomain are required for specific high-affinity DNA binding of the Pit1-responsive elements. Pit1 protein is able to bind as a monomer in solution to the consensus (A/T)(A/T)TATNCAT site, where N may be any nucleotide; in most cases, however, Pit1 binds DNA as a dimer.41 Analysis of data derived from a cocrystal study of the Pit1 protein and the PRL proximal promoter Pit1-binding element reveals that the Pit1 protein binds to DNA in a parallel dimer form.42,43 Pit1 protein wraps around the DNA molecule, with the POU-specific domain and the POU homeodomain binding to the DNA molecule in a perpendicular angle in opposite orientation. The POU-specific domain of one Pit1 molecule interacts with the C terminus of the POU homeodomain of the other Pit1 molecule in a dual composition. In addition, the spacing between the DNA contacts made by the POU-specific domain and the POU homeodomain of each monomer is critical. Compared to the Pit1 binding site in the PRL minimum promoter sequences, two additional base pairs spacing are needed to direct restricted GH gene transcription based on elements of two Pit1 binding sites on the proximal promoter of rat GH locus.44
This dimerization interface is a “hot spot” for debilitating mutations. Additional mutations, like in the Pit1Snell mice, a G-to-T mutation results (W261C) in the third helix of the POU homeodomain, eliminating its DNA-binding ability by altering the contact point of the mutant gene product with the major groove of the responsive elements, causing a dwarf phenotype in an autosomal-recessive fashion. Similarly, several mutations observed in human cases could affect the stability and specificity of this protein-DNA interface.45,46
As a transcription factor, Pit1 exerts its effects as a component of a transcriptional complex regulated by coactivator and repressor elements. The Pit1 POU domain can associate with coactivator complex of CBP/p300 and P/CAF, both of which possess histone acetylase activity. N-CoR, acting as a corepressor, can bind to the homeodomain of Pit1 and actively suppress transactivation by Pit1; this suppression depends on Sin3, SAP30, and histone deacetylase. Thus the transcriptional activity of Pit1 may be regulated by the competing binding of complexes mediating either acetylation or deacetylation events, resulting in activation or repression, respectively.47
In addition to Pit1, the determination of individual pituitary cell types may require other molecules. The estrogen receptor has been implicated in synergistic activation of the PRL gene.48,49 Members of the ETS family of transcription factors can bind to Pit1 binding sites in the PRL promoter and mediate signals from growth factors and the Ras/mitogen-activated protein kinase pathway.50 The transcription factor GATA2 appears to be required for the formation of both thyrotrope and gonadotrope cells; the presence of Pit1 represses the gonadotropic phenotype and promotes the thyrotrope phenotype. Pit1 can inhibit binding of GATA2 to cognate DNA sites important for generation of the gonadotrope phenotype. In contrast, Pit1 leads to synergistic activation with GATA2 on promoters that contain both Pit1 and GATA2 sites.51
PROP1 GENE
Prop1 (Prophet of Pit-1) is a homeodomain-containing transcription factor that is capable of binding to its cognate DNA site and activating its target genes. The expression pattern of the Prop1 gene has been examined in mice and is detected only in Rathke’s pouch. Prop1 expression is detected initially at e10 in the mouse, when the structure of Rathke’s pouch has been established. The expression initially is observed dorsally but subsequently involves most cells in Rathke’s pouch. Expression of Prop1 reaches a maximum level of intensity at e12 in Rathke’s pouch, with the signal diminishing by e14.552 (see Fig. 8-1). It has been shown recently that Notch signaling is required for maintaining high levels of Prop1 expression at e12.5, which is mediated by Rbp-J protein bound to the evolutionary conserved site within the first intron of the Prop1 gene.53 Expression of the Prop1 gene is required for activation of the downstream Pit1 gene.54,55 The integrity of Prop1 is necessary for full-scale manifestation of pituitary gonadotrope cells, as well as the generation of somatotrope, lactotrope, and thyrotrope cells (see Table 8-2). Mutations in the Prop1 gene have been identified as the leading cause of familial CPHD, resulting in short stature as a consequence.
Animal Model
The Prop1 gene was initially identified by a positional cloning strategy in the naturally occurring Ames mouse mutant. The mutant Prop1 allele at the Prop1Ames locus harbors a point mutation that results in a single amino acid substitution (S83P) in the second helix of the homeodomain, causing altered progression of nascent pituitary gland and subsequent failed expression of Pit1.52 Phenotypes of the Prop1Ames mice are transmitted in an autosomal-recessive fashion; heterozygous mutant mice are normal. Homozygous mutant mice are born grossly normal but develop a proportional dwarfism by the time of weaning.56 The adult mutant mice are about half the size of the wild-type animals. The Prop1Ames mutation caused dysmorphogenesis of Rathke’s pouch at e12.5, with convolution of the lumen and a failure of expression of the Pit1 lineage. The appearance of gonadotrope was delayed, but corticotrope appeared as expected. In contrast to the complete absence of somatotrope, lactotrope, and thyrotrope cells in the Pit1Snell mouse, the Prop1Ames mouse pituitary gland contains a small number (<1%)54,57 of the normal complement of somatotrope cells, as well as a few lactotrope and thyrotrope cells.57 Prop1Ames dwarf mice live twice as long as their wild-type littermates.58
Related Diseases
The human Prop1 coding region has three exons separated by two introns and maps to chromosome 5q34. The Prop1 gene encodes a polypeptide of 226 amino acids and contains a short N terminus, a 60-amino acid homeodomain, and a transactivating C terminus. Compared to the mouse homologue, the human Prop1 homeodomain is highly conserved, with only two amino acid substitutions.
Initial reports identified mutations in the human Prop1 gene in patients with short stature in several families. Direct sequencing of polymerase chain reaction (PCR) products of the Prop1 gene revealed that all the affected patients were harboring mutations in both alleles of the Prop1 gene, and their parents were heterozygous for the respective mutations, suggesting that the mutations in the Prop1 gene act in an autosomal-recessive manner, causing CPHD in these patients. All of the affected individuals in this study failed to respond to GHRH, thyrotropin-releasing hormone, and LH-releasing hormone stimulation, suggesting a defect in hormone-secreting cells of the pituitary gland.59 Subsequent reports have revealed that Prop1 mutation is a common cause of familial CPHD. These alternations in the Prop1 gene range from point mutation to deletions, affecting structure and integrity in the homeodomain of the Prop1 gene. A 2-bp A301G302 deletion, leading to a frame-shift and the loss of DNA-binding homeodomain and C-terminal transactivation domain of the Prop1 gene product, is the most frequently encountered mutation among these Prop1 patients, representing a mutational “hot spot.”60 Individuals with various Prop1 mutations invariably display severe deficiencies for pituitary gonadotropins in addition to the defects of GH, PRL, and TSH levels. In human cases with Prop1 mutations, many adult patients express ACTH at a normal level; however, there are reported cases with a late onset of corticotropin deficiency (see Table 8-2). The expression of ACTH phenotypes is highly heterogeneous; differences in genetic background in these patients may contribute to the discrepancy of this phenotype.61,62
Mechanism
The Prop1 gene product exerts its actions through binding to the responsive elements of target genes, with the helix-turn-helix motif of the homeodomain providing the contact point for protein-DNA interactions. The fact that most of the naturally occurring mutations of the Prop1 gene are located in the homeodomain suggests that the Prop1 homeodomain is critical for Prop1 function.
In Prop1Ames mice, examination of the mutant Rathke’s pouch revealed severe dysmorphogenesis, but the pituitary precursor cells were generated. The precursor cells of Rathke’s pouch failed to migrate to form the nascent pituitary gland, leading to an expansion of the luminal structure and lack of expression of a late pituitary differentiation marker, the Pit1 gene. However, proliferation of the mutant precursor cells in the Ames mice continued, resulting in normal-sized pituitary glands.52,63 In addition to Pit1, both Wnt and Notch pathways are affected in the Prop1Ames mice.64,65 Later, persistent expression of Prop1 under control of the αGSU promoter caused decreased gonadotrope differentiation and increased adenomatous hyperplasia,66,67 indicating that properly extinguishing Prop1 also may be an important later step in paired-like homeodomain-mediated organogenesis.
Prop1 can bind to its site and activate target genes via the C-terminal transactivation domain, whereas the N terminus and the homeodomain of Prop1 possess repression function,52,68 suggesting that Prop1 can act as a transcriptional activator as well as a repressor. Recent studies of the pituitary-specific inactivation of the β-catenin gene reveal that a Prop1/β-catenin complex acts as transcriptional activator for Pit1 and as a repressor for Hesx1, depending on the associated co-factors.69
Phenotypic comparisons have been made between the Prop1-defective patient and the Prop1-mutant Prop1Ames mouse. Deficiencies of GH, PRL, and TSH are consistently observed in both species. All the patients with Prop1 mutations eventually develop gonadotropin deficiency in their adult lives. In the Prop1Ames mice, the expression of gonadotropin is observed at birth, but the level of expression of the gonadotropin is reduced to one quarter of that of the wild-type animals.52 The expression of ACTH is apparent during development in the Prop1Ames mouse pituitary, and the level of ACTH in the blood is normal in adults. In human Prop1 patients, cortisol levels are normal at birth, but some patients develop cortisol deficiency later in life.70–72 The Prop1 mutation may affect all the major cell types in the anterior pituitary gland, including the gonadotrope and the corticotrope (see Table 8-2).
HESX1 GENE
Hesx1 (homeodomain gene expressed in ES cells) is a paired-class homeodomain transcription factor that is capable of binding to its cognate DNA site and regulating its target genes. Mutations in the Hesx1 gene have been identified in septo-optic dysplasia and CPHD. In mice, the earliest expression of the Hesx1 gene can be detected at the embryonic stem cell stage. High levels of expression can be detected in the ectoderm, subsequently at the anterior extreme of the rostral neural folds, and finally restricted to the ventral diencephalon and to the thickened layer of oral ectoderm, which will give rise to Rathke’s pouch at e9.0 in the mouse.73,74 Hesx1 gene expression can be observed for 2 more days but only in Rathke’s pouch, with diminishing intensity at a time that coincides with the rise of Prop1 gene expression (Fig. 8-2; also see Fig. 8-1). In humans, strong expression of Hesx1 in Rathke’s pouch can be detected in a 7-week-old embryo. Hesx1 is the earliest molecular marker for the definitive pituitary primordium.
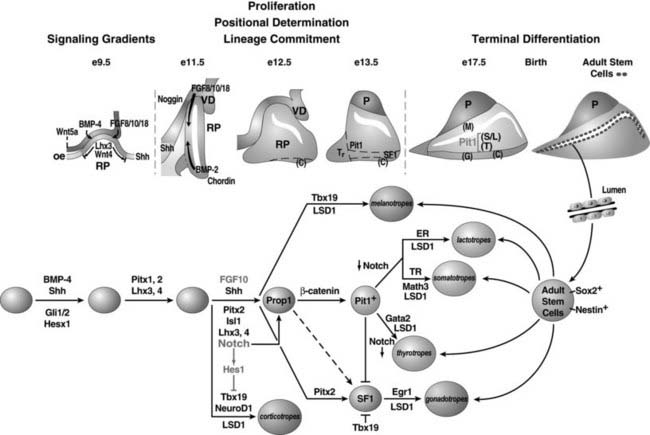
FIGURE 8-2. Ontogeny of signaling molecules and selected transcriptional factors during mouse pituitary organogenesis. Ventral diencephalon, which expresses BMP4, FGF8/10/18, and Wnt5, makes direct contact with oral ectoderm and induces the formation of Rathke’s pouch. Shh is expressed throughout the oral ectoderm, except in Rathke’s pouch, creating a boundary between two ectodermal domains of Shh-expressing and nonexpressing cells. The opposing dorsal BMP4/FGF and ventral BMP2/Shh gradients convey proliferative and positional cues by regulating combinatorial patterns of transcription factor gene expression. Pit1 is induced at e13.5 in the caudomedial region of the pituitary gland, which ultimately gives rise to somatotropes (S), lactotropes (L), and thyrotropes (T). Rostral tip thyrotropes (Tr) are Pit1 independent. Corticotropes (C) and gonadotropes (G) are differentiated in the most ventral part of the gland. The dorsal region of Rathke’s pouch becomes the intermediate lobe, containing melanotropes (M). The infundibulum grows downward and eventually becomes the posterior lobe (P). A number of transcription factors and cofactors regulating the lineage commitment and terminal differentiation of distinct cell types are illustrated in a genetic pathway.
(Modified from Zhu X et al: Signaling and epigenetic regulation of pituitary development. Curr Opin Cell Biol 19(6):605–611, 2007.)
Animal Model
The mouse Hesx1 gene is located on chromosome 14, and targeted deletion of Hesx1 resulted in mice that exhibited variable anterior central nervous system defects with reduced prosencephalon and defective olfactory development.75 Hesx1 mutants also have defects in the pituitary gland, with bifurcations in Rathke’s pouch in most cases. By e12.5, multiple oral ectoderm invaginations reflecting pituitary glands are observed in most Hesx1 embryos. Between e13.5 and e15.5, Hesx1 mutants are characterized by a dramatic cellular over-proliferation of all the hormone-producing cell types, leading to a failure of the underlying mesenchyme to condense and form the sphenoid cartilage that separates the pituitary from the oral cavity. In the late stages of pituitary development, the terminal differentiation of the hormone-producing cell types appear normal in most Hesx1 mutants, with overexpression of αGSU, TSHβ, GH, POMC, and Pit1 by e16.5. Earlier in development, there is a delay in the onset of POMC expression both in Rathke’s pouch and in the developing hypothalamus at e12.5, and there also appears to be a dual induction of αGSU expression on both the rostral and caudal sides of Rathke’s pouch. Strikingly, in occasional Hesx1 gene–deleted mice, the initial thickening of oral ectoderm and minimal activation of Lhx3 are observed at e12.5, but the embryos exhibit a complete arrest of pituitary development, and the pituitary gland is absent by e18.5. The discrepancy of incomplete phenotype penetrance in Hesx1 mutants is likely influenced by the actions of the linked modifier genes.76,77
Related Diseases
The human Hesx1 gene contains four exons separated by three introns, and it maps to chromosome 3p21. The Hesx1 gene encodes a highly conserved polypeptide of 185 amino acids with a 60-amino acid homeodomain at its C terminus. Initial analyses of the Hesx1 mutations carried out in kindreds with septo-optic dysplasia identified a nucleotide transition that resulted in the substitution of R160C (in the third helix of the homeodomain) in two children with CPHD born to a highly consanguineous family. Magnetic resonance imaging revealed an ectopic/undescended posterior pituitary associated with a hypoplastic anterior lobe in these two affected siblings.75 None of the heterozygote parents exhibited features of septo-optic dysplasia, consistent with an autosomal-recessive inheritance. Additional mutations (e.g., Q6H, S170L, T181A, I26T, and 306/307InsAG-X) have been found in the coding region of the Hesx1 gene and are associated with variable phenotypes, including hypopituitarism, ranging from isolated GH deficiency to CPHD. It is clear from these reported cases that mutation in the Hesx1 gene can cause pituitary hormone deficiency with variable phenotypes and with incomplete penetrance.78,79
Mechanism
The Hesx1 gene product can bind to either dimer or monomer DNA sites with high affinity in transient transfection assays.80,81 Modular structure analysis revealed that in addition to the DNA-binding homeodomain, Hesx1 contains two sequences in the N terminus; one is similar to the eh1 motif found in Drosophila engrailed, and one is similar to the WRPW motif found in several helix-loop-helix proteins, both of which are capable of recruiting the Groucho class of corepressors.82,83 Both the N-terminal and homeodomain regions of Hesx1 can independently act as repressors. Hesx1 is a strong transcriptional repressor that acts by recruiting the mSin3A/B, HDACs 1 and 2, and the Brg1 complexes to its homeodomain and the TLE corepressor to its eh1 domain. The strong association between Tle1 and Hesx1 is mediated by a highly conserved helical motif (FXLXXIL) present in the Hesx1 N terminus, which can also be found in Nkx, Six, and certain Pax homeodomain factors’ family members.84 These recruitments are required and sufficient for the repressive actions of Hesx1 in vivo. Forced persistent expression of Hesx1 and Tle1 resulted in the loss of the Pit1 lineage and a Prop1Ames-like dysmorphogenesis, while the expression of Prop1 and POMC remained. The mutation in human Hesx1 (R160C) has a dominant negative effect both in vitro and in vivo. This dominant negative activity requires the eh1 repression domain, which is also required for full-length recombinant Hesx1 dimerization in solution. This dominant transcription repressor activity may help to explain the heterozygous phenotypes observed in Hesx1 patients.80 Recent identification of a homozygous mutation in the eh1 motif (I26T) in a patient with CPHD has further underlined that Tle association is an integral mechanism for Hesx1 function in vivo.85
Hesx1 and Prop1 share a conserved DNA-recognition site. The repression domain in Hesx1 can suppress the transcription activation activity of Prop1. The Hesx1 repressor can heterodimerize with Prop1 and can bind to the palindromic site as homodimers or heterodimers, with Prop1 acting as an activator and Hesx1 as a repressor, to inhibit Prop1 activation function. The expression of Prop1 is elevated in Hesx1-mutant mice, suggesting not only that Hesx1 can repress Prop1 activation function but also that it is required for proper Prop1 expression.76 Forced early expression of Prop1 to the uncommitted oral ectoderm blocks the formation of Rathke’s pouch, which results in absence of the anterior pituitary gland with no initial induction of Lhx3 expression, demonstrating that premature expression of Prop1 can block the pituitary organogenesis that phenocopies the effects of Hesx1-gene deletion,15 in contrast to the Hesx1/Tle1 transgenic mouse with a Prop1Ames-like phenotype, suggesting that the antagonistic repressor complex can suppress Prop1 activation of expression.76 The sequential repression and activation of a common set of regulatory genes may prove to be an underlying strategy in the temporal code of pituitary organ development, with initial repression required for organ commitment and proliferation and subsequent activation required for commitment of specific cell lineages.69
LHX3 AND LHX4 GENES
Lhx3 (LIM homeo box gene 3) is a LIM-type homeodomain transcription factor. In addition to a C-terminus homeodomain, Lhx3 contains two tandem repeats of LIM zinc-binding motifs, each composed of 50 to 60 amino acids with a conserved pattern of cysteine and histidine residues that form a pair of zinc fingers, separated by a linker of 2 amino acids. Expression analysis revealed that mouse Lhx3 mRNA can be detected in the developing nervous system and accumulates in Rathke’s pouch beginning at e9.5 (see Fig. 8-1). Lhx3 remains expressed in the entire pouch, and its expression is maintained through e15.5; the expression is particularly strong in the anterior and intermediate lobes of the adult pituitary. In addition, Lhx3 is expressed bilaterally along the spinal cord and the hindbrain at early stages of development.86
Structurally, Lhx4 (LIM homeodomain gene 4) is closely related to Lhx3. The Lhx gene gamily consists of at least 12 members; many of them are expressed in the pituitary during development, including Isl1, Isl2, Lhx2, Lhx3, and Lhx4. Lhx3 and Lhx4 have been genetically defined as required elements for both the early stages of pituitary determination and the later differentiation of pituitary cell types. By in-situ hybridization, the Lhx4 gene is found to be expressed transiently in ventrolateral regions of the neural tube and the hindbrain of the developing mouse. During pituitary development, Lhx4 is expressed throughout the invaginating Rathke’s pouch at e9.5. At e12.5, Lhx4 expression becomes restricted to the future anterior lobe of the pituitary gland, and by e15.5, Lhx4 expression diminishes. In the adult pituitary, Lhx4 is found in the anterior and intermediate lobes at a much lower level than that of Lhx3.87
Animal Model
Employing a reverse genetic approach, mice with a targeted disruption in the Lhx3 gene were generated. Mice heterozygous for the mutation are apparently normal and fertile, whereas homozygous individuals are stillborn or expire within 24 hours of birth. In these homozygous mice, the hindbrain, spinal cord, and pineal gland are grossly normal, as is the posterior lobe of the pituitary, but the anterior and intermediate lobes of the pituitary are absent. During embryonic development, the mutant animal exhibits a lack of growth in Rathke’s pouch, and pituitary-gland development does not progress beyond the Rathke’s pouch stage. With the exception of residual corticotrope, other anterior pituitary cell types are absent, indicating that Lhx3 is required for the appearance of the somatotrope, lactotrope, thyrotrope, and gonadotrope cell types.88
Mice homozygous for the targeted deletion of the Lhx4 gene exhibit an early postnatal death due to a failure of pulmonary maturation.87 Lhx4-deleted mice have a well-formed Rathke’s pouch but display incomplete pituitary development following this stage, and the differentiation of pituitary cell types is perturbed. Consequently, by e12.5, there exists a miniature Rathke’s pouch, and by e14.5, the nascent pituitary structure has progressed to a larger pouch, but the anterior lobe is discernible only as a slight thickening in the ventral region. This hypocellularity of the anterior lobe is caused by failure of pituitary precursor cells to survive; large numbers of apoptotic cells are evident throughout the pituitary primordia of Lhx4-mutant mice at e12.5.89 In later gestation stages, Rathke’s pouch is hypoplastic, with an enlarged lumen resulting from reduced proliferation of the precursors, and the anterior lobe of the pituitary is reduced in size. Expression analyses have revealed residual amounts of LH- and GNRHR-positive cells at e18.5. Thus, Lhx4 is not required for specification of gonadotrope cells, but it does support the expansion of the cell population. Similarly, all five anterior pituitary–specific cell lineages are present in the Lhx4-mutant pituitary but in dramatically reduced numbers. By contrast, the intermediate-lobe melanotrope cells are undisturbed.
Mice with double deletion of Lhx3 and Lhx4 demonstrated that both genes direct formation of the pituitary gland.90 The early formation of the Rathke’s pouch rudiment from pituitary primordium does not depend entirely on the function of either Lhx3 or Lhx4 alone, but together these genes redundantly control the formation of the definitive pouch. Lhx3 also controls a subsequent step of pituitary fate commitment, and in these early stages, Lhx4 appears to act upstream of the Lhx3 and Isl1 genes and is required for expansion of Rathke’s pouch. Therefore, Lhx3 and Lhx4 dictate pituitary gland identity by controlling decision points of organogenesis and regulation of the proliferation and differentiation of pituitary-specific cell lineages.
Related Diseases
Human Lhx3 shares a high degree of homology with its mouse orthologue, exhibiting 94% identity at the amino acid level. Lhx3 is located on human chromosome 9q34 and spans a genomic fragment of at least 6 kb that includes 6 exons.91,92 In a candidate-gene screen based on pituitary phenotypes observed in a recessive lethal mutation in mice, two mutations in the Lhx3 gene were identified in two unrelated consanguineous pedigrees that display CPHD.93 In one family, affected individuals are homozygous for a Y116C mutation located in the highly conserved LIM2 domain of Lhx3, a domain critical for protein-protein interactions. In the second family, affected individuals are homozygous for a 23-base-pair deletion in an intragenic region, predicting a severely truncated protein that lacks the entire homeodomain and rendering it incapable of DNA binding. Lhx3-defective patients have deficiencies in GH, TSH, PRL, FSHβ, and LHβ, but they display intact levels of ACTH, similar to the endocrine profiles observed in Prop1 patients (see Table 8-2). In addition, these Lhx3-defective patients displayed a rigid cervical spine that restricted their head rotation. More recently, novel 6 mutations have been found in the coding region of the Lhx3 gene.94,95 All of them are associated with variable phenotype of hypopituitarism. Lhx3 mutations are a rare cause of CPHD involving deficiencies for GH, prolactin, TSH, and LH/FSH in all patients. Whereas most patients have a severe hormone deficiency manifesting after birth, milder forms can be observed, and limited neck rotation is not a universal feature of patients with Lhx3 mutations.
The human Lhx4 gene encodes a 390-amino acid protein that contains two LIM domains and a homeodomain that shares 99% sequence identity with its mouse orthologue. Genomic analysis revealed that the human Lhx4 gene contains 6 exons and is mapped to chromosome 1q25.96 In a large consanguineous pedigree of three generations, a G-to-C substitution in the intron preceding exon 5 of Lhx4 generates a mutant protein with perturbed homeodomain, which affects its DNA-binding function. Patients with this disease have short stature with CPHD, which affects GH, thyroxine, and cortisol, as well as cerebellar defects and abnormalities of the sella turcica. This mutant allele is transmitted in a dominant fashion, affecting only the maternal side of the kindred with a high phenotypic penetrance.96 More recently, three novel mutations in the Lhx4 gene have been mapped.97 All of them affect the DNA-binding domain, and all of them are associated with CPHD.
Mechanism
LIM homeodomain proteins are transcription factors and exert their effects by regulating target gene expression. Lhx3 binds with high affinity to AT-rich DNA sequences (including minor groove interaction) and bends the DNA molecule to an angle of 62 degrees in a model system.92 Lhx3 can activate the regulatory regions of pituitary genes, including αGSU, PRL, TSHβ, and Pit1. Lhx3 expression is partially regulated by the Lhx4 gene during pituitary development. At e12.5, only a few cells express Lhx3 in the dorsal-most aspect of the pouch in the Lhx4 mutants. However, the normal pattern of Lhx3 expression, including the dorsal-ventral gradient, is established in Lhx4 mutants by e14.5.89
Genetic analysis revealed that Lhx4 interacts with Prop1 to stimulate anterior pituitary lobe expansion. Neither gene is essential for initiating corticotrope specification. However, no POMC or αGSU expression is detected in double-mutant mice at e14.5, suggesting that Prop1 and Lhx4 have overlapping roles in corticotrope and gonadotrope development.89 In Hesx1-deleted mutants, the domains of Lhx3 and Prop1 expression are increased, as well as those of FGF8 and FGF10 in the infundibulum, which become expanded rostrally.76 These findings indicate that Hesx1 is required for maintaining the proper expression of FGFs, consistent with the notion that Lhx3 expression can be regulated by FGF signaling.
TBX19 GENE
Tbx19 is a T-box transcription factor family member (the T-box in the mouse T [Brachyury] gene) that encodes a 448-amino acid protein.98 Functional identification of Tbx19 was established after the observation of elements in a critical cis-acting sequence in the POMC promoter. Transcripts of Tbx19 can be found only in the anterior and intermediate pituitary and brain (see Fig. 8-1); Tbx19 is specifically required for continued POMC transcription.99,100
Animal Model
Mice with targeted disruption of the Tbx19 gene have been generated. Mice heterozygous for the mutation are apparently normal. Adult mice homozygous for the mutation have very few ACTH-positive cells in the pituitary, although initial expression of the POMC gene is undisturbed at the Rathke’s pouch stages. These cells are born in normal quantities in mutants but are lost or fail to expand appropriately, suggesting Tbx19 is not required for corticotrope cell commitment but is later important for POMC lineage differentiation. The intermediate-lobe melanotropes in mutant mice are populated by gonadotrope and some Pit1-independent thyrotrope, also indicating that Tbx19 normally represses pituitary gonadotrope differentiation.101,102
Related Diseases
The human Tbx19 gene shares 94% amino acid identity with that of mouse Tbx19 and maps to chromosome 1q23-q24. Several cases of isolated ACTH deficiency were identified with a nonsense mutation C-to-T transition in exon 6 in Tbx19, resulting in a truncated gene product (R286X).103 The transmission of this mutation appears to be recessive. In another case of isolated ACTH deficiency, a heterozygous C-to-T transition in exon 2 of the Tbx19 gene was identified, resulting in a conserved amino acid S128F mutation, suggesting a dominant negative inheritance.99 More recent study revealed new mutation in the Tbx19 gene (missense M86R) that did not affect monomer DNA-binding activity per se, but it impaired DNA binding with other DNA-bound proteins, including itself (homodimers) and Pitx1, resulting in congenital isolated ACTH deficiency.104 Additional mutations in the Tbx19 gene have been shown to be a cause of neonatal death due to neonatal-onset isolated ACTH deficiency.103 Tbx19 defects result in POMC deficiency in both humans and mice, establishing Tbx19 as the gene required for effective POMC expression in vivo.101,102
Mechanism
Tbx19 is a transcriptional regulator, recognizing target genes through its T-box DNA-binding domain. In response to signals elicited by the hypothalamic hormone corticotrope-releasing hormone, Tbx19 functions as an activator of transcription by recruiting SRC/p160 coactivators to its cognate DNA target in the POMC promoter.105 Tbx19 can synergize with orphan nuclear receptor NGFI-B, serving as part of the transcription regulatory complex on the POMC promoter in response to hormonal stimulation.106 Transgenic expression of Tbx19 in non-POMC-producing regions of the pituitary gland can cause ectopic POMC expression,99 and Tbx19 is an inhibitor of αGSU expression in rostral tip cells, gonadotrope, and thyrotrope, and of TSHβ production in caudomedial thyrotrope.100 Tbx19 deficiency is permissive for transdifferentiation of cells normally destined to be corticotrope and melanotrope into alternative cell fates, namely, gonadotrope and rostral tip thyrotrope, suggesting a determinative role of Tbx19 in cell lineage specification. Tbx19 defects have no effect on differentiation of Pit1-dependent cell lineages (see Figs. 8-1 and 8-2, and Table 8-2).101,102
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


