in a different channel. The interference is corrected by applying fluorescence compensation based on data from single-stained samples. This is usually done using cells or beads before or during the data acquisition phase. However, modern FCM data analysis software also allows collection of uncompensated data and applying compensation during analysis. Before data acquisition, standard reference particles (fluorescent microspheres) should be used to adjust the PMT voltage settings so that the beads fall in approximately the same location or the same “target channels,” predetermined for each fluorochrome.
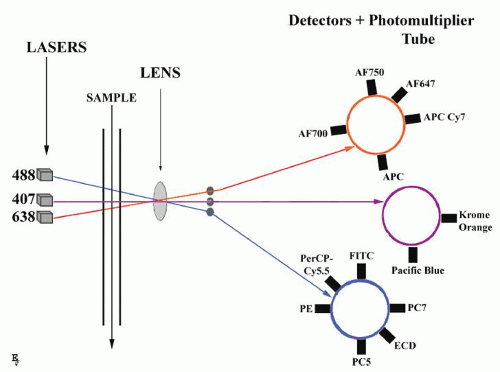 FIGURE 2.1. Principles of multicolor flow cytometry. A single cell suspension is hydrodynamically focused with sheath fluid to intersect lasers (three-laser system is shown). Fluorescence signals are collected by multiple fluorescence emission detectors, separate for every laser. Examples of fluorochromes detected by different lasers are given according to Table 2.1. Detected signals are amplified by photomultiplier tubes and converted to digital form for analysis. |
TABLE 2.1 TABLE OF FLUOROCHROMES COMMONLY USED IN CLINICAL FLOW CYTOMETRY | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A droplet generator
A droplet charging and deflecting system
A collection component
The electronic circuitry for coordinating the timing and generation of droplet-charging pulses
cell lines (hybridomas) that produce an antibody of unique specificity, avidity, and isotype can be established. In the early days of the application of MAbs to immunology, many laboratories were immunizing mice with leukocytes. The obtained hybridomas produced many antibodies that reacted with leukocytes, but the identities of the molecular targets were not known. The reactivity spectrum of the antibody could be described by staining multiple different cell types, and in most cases the target antigen could be isolated by immunoprecipitation or Western blotting and its molecular weight and other structural characteristics determined.
TABLE 2.2 LIST OF CD ANTIGENS MOST COMMONLY USED IN FLOW CYTOMETRY IMMUNOPHENOTYPING OF HEMATOLOGIC SAMPLES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mature lymphocytes are characterized by low side scatter and strong CD45 expression (lymph region, Fig. 2.2 plot B).
Monocytes have higher SS and strong CD45 expression (monocyte region, Fig. 2.2 plot B).
Erythropoietic precursors are CD45 negative and have low SS (CD45- ery region, Fig. 2.2 plot B).
Granulopoietic precursors and granulocytes are weakly CD45 positive and have high SS (CD45 dim, gran region, Fig. 2.2 plot B).
Early hematopoietic precursors of various lineages, including CD34+ stem cells, are characterized by low CD45 expression and low SS (blast region, Fig. 2.2, plot B).
Histograms (for one parameter), where relative fluorescence or scatter is on the x-axis and the number of events with given characteristics on the y-axis
Two-parameter dot plots, where each signal is visualized by one dot and given a parameter on the x– and y-axes; various cell populations can be then “painted” with different colors
Density plots, where hotspots indicate large numbers of events resulting from discreet population of cells and colors can give the graph a three-dimensional feel
Contour diagrams, where joined lines represent similar numbers of cells
Demographic identification of patient
Identification of the hospital or division sending the sample
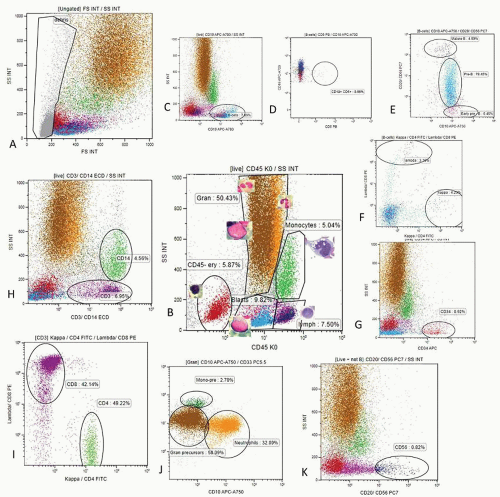
FIGURE 2.2. Bone marrow mapping with polychromatic flow cytometry. Reactive bone marrow sample from a young patient was analyzed with a screening ten-color 14 MAb panel on a Navios flow cytometer and Kaluza software (Beckman Coulter). The MAb panel consisted of kappa+CD4 FITC/Lambda+CD8-PE/CD3 + CD14 ECD/CD34 APC/CD20+CD56-PC7/CD10-APC-A750/CD19-APC-A700/CD33 PC5.5/CD5-Pac Blue/CD45 Krom Orange. Analysis starts with the creating of the “live cells” gate by removal of dead cells, erythrocyte, and platelet aggregates on FS/SS plot (A). A CD45/SS plot is created within the live cell gate (B). Regions for lymphocytes (CD45bright/low SS), monocytes (CD45 bright/high SS), granulopoietic cells (CD45 dim/high SS), CD45dim/low SS blasts, and CD45-low SS erythropoietic cells are determined. The B-cell gate is created from the live cell gate on the CD19/SC plot (C). Presence of CD5 positive B-cells is investigated using a CD5/CD19 plot (D). The presence of CD10+ B-cells is looked for by analysis of CD20 and CD10 expression within the B-cell gate (E). In this patient, no CD5+ B-cells were detected but a significant fraction of B-cells showed B-precursor immunophenotype with normal maturation pattern (E). If a CD5+ or CD10+ B-cell population is present, a new gate can be created within plot D or E. B-cell clonality is analyzed within the B-cell gate (F). In this patient most B-cells are negative for light chain expression, consistent with B-cell precursors. Note that most of CD10+/CD20 dim B-cell precursors (cyan dots) fall into the blast gate in the CD45/SS plot (B). Kappa and lambda positive B-cells have normal kappa to lambda ratio. If CD5 and/or aberrant CD10+ B-cells were present, clonality of B-cells would be analyzed within the specific CD5+/CD19+ or CD10+/CD19+ gate. The fraction of CD34+ cells (red dots) is estimated within the live cell gate on the CD34/SS plot (G). If increased numbers of CD34+ cells are found, they are further analyzed for CD33, CD19, and CD10 expression. CD3+ T-cell and CD14+ monocyte gates are created on the CD45/CD3+CD14 plot within the live cell gate (H). Fractions of CD4+ (violet dots) and CD8+ T-cells (light green dots) are estimated within the CD3+ gate (I). CD4/CD8 ration was normal (1.16). Granulopoietic cells are analyzed on CD33/CD10 plot within the “Gran” gate and fractions of mature neutrophils (CD33+CD10, orange dots) and granulopoietic precursors (CD33+ CD10, brown dots) are estimated (J). CD14-CD33bright monocytic precursors can also be enumerated (green dots). Finally the fraction of CD56+ NK cells (dark blue dots) can be evaluated on a CD20+56/SS plot using the Boolean gate of live cells + non-B-cells to exclude CD20+ B-cell from analysis (K). Various cell populations are back-gated and visualized on both FS/SS and CD45/SS plots (A and B).
Type of specimen (bone marrow aspirate, peripheral blood, other biologic fluids)
Timing of observation (first diagnosis or follow-up)
Diagnostic hypothesis made by the sender
List of antigens and type of immunofluorescence analysis carried out
Absolute number of cells in the sample
Quality of the sample, in terms of viability
General description of the gating procedure
Immunophenotype of abnormal cells present in the sample
Description of other (normal) cells
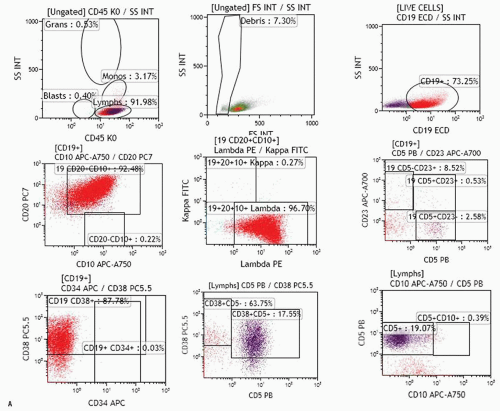
FIGURE 2.3. A. Examples of analysis of B-cell compartment in bone marrow samples. Ten-color MAb panel, Navios flow cytometer, and Kaluza software (Beckman Coulter) were applied. Panel consists of Kappa-FITC/Lambda-PE/CD19 ECD/CD34-APC/CD10-APC-A750/CD23-APC-A700/CD20-PC7/CD38-PC5.5/CD5 Pc Blue/CD45-Krom Orange. The live cell gate is created and fractions of lymphocytes, granulocytes, monocytes, and the like are evaluated as shown in Figure 2.2. The B-cell gate is created on a CD19/SS plot and expression of CD5, CD23, and CD10 is analyzed within the B-cell population. Kappa and lambda light chain expression is analyzed within total B-cell, CD5+ B-cell, or CD10+ B-cells as appropriate. Expression of CD34 and CD38 within the C19+ B-cell population can also be analyzed (see Fig. 2.3B). The fraction of plasma cells can be estimated using CD38 bright expression and high SS on the CD38/SS plot (not shown). Upper row: population of B-cells with B-CLL/small lymphocytic lymphoma-related phenotype (CD19+, CD5+, CD23+, CD20 dim, kappa dim, CD10-) consistent with bone marrow involvement in a patient who was diagnosed with small lymphocytic lymphoma in a lymph node biopsy and had no peripheral lymphocytosis. Bone marrow biopsy showed rare nodular lymphoid infiltrates. Middle row: CD5-CD10-CD23- lambda+ B-cell population in a patient with Waldenström macroglobulinemia. Lower row: population of CD19+ CD10+ B-cells strongly expressing CD20 and kappa in a patient with bone marrow involvement by a follicular lymphoma. B. Examples of analysis of B-cell compartment in a lymph node cell suspension. Ten-color MAb panel, Navios flow cytometer, and Kaluza software (Beckman Coulter) were applied. Panel consists of Kappa-FITC/Lambda-PE/CD19 ECD/CD34-APC/CD10-APC-A750/CD23-APC-A700/CD20-PC7/CD38-PC5.5/CD5 Pc Blue/CD45-Krom Orange. The live cell gate is created and fractions of lymphocytes, granulocytes, monocytes, and the like are evaluated as shown in Figure 2.2. The B-cell gate is created on the CD19/SS plot and expression of CD5, CD23, and CD10 is analyzed within the B-cell population. Most B-cells were positive for CD20, CD38, and CD10, and showed monotypic lambda expression.
Diagnostic conclusions
methodology, interlaboratory comparison, or verification with specimens obtained from patients with a confirmed diagnosis. A minimum of 10 to 20 samples (10 normal, 10 abnormal) is recommended for accuracy assessment. The acceptance criteria will also be variable depending on the required degree of accuracy for the intended use, nevertheless should be clearly defined for each assay. Ninety percent, or greater, agreement between methods is generally required for accuracy.
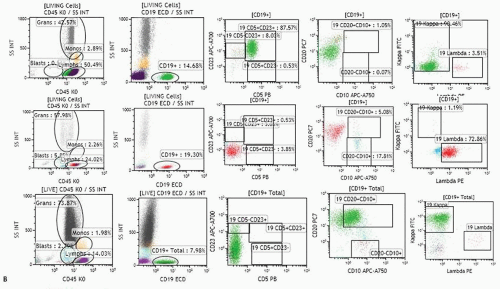 FIGURE 2.3. (Continued) |
TABLE 2.3 EXAMPLES OF 10-COLOR FLOW CYTOMETRY PANELSa IN IMMUNOPHENOTYPING OF LEUKEMIA AND LYMPHOMA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
patterns. Following reports by Terstappen et al.,29,30 and 31 several groups provided descriptions of clearly delineated differentiation stages of various hematopoietic cell lineages.32,33,34,35 and 36,37,38 A detailed review of all available data is beyond the scope of this chapter; a summary of the most important and well-established issues is provided below.
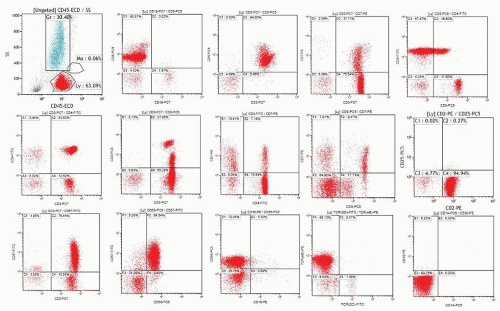 FIGURE 2.4. Example of aberrant T-cell population detected in peripheral blood of a patient with lymphocytosis. Five-color MAb panel and FC500 flow cytometer (Beckman Coulter) were used. Analysis shows that 63% of blood cells were lymphocytes (Ly, red dots on upper left plot). Analysis was performed within lymphocyte gate. Analysis revealed an aberrant population of CD3+ T-cells (75% of lymphocytes) that lack CD7 are positive for CD5 and CD4 with partial co-expression of CD8 (upper row). Small populations of normal CD4+CD7+ and CD8+CD7+ T-cells are also noted (7% and 9% of lymphocytes, respectively, middle row). All T-cells were positive for CD2 and negative for CD25 (left middle row). Further analysis that showed that the aberrant T-cell population was positive for NK-cell-associated antigens CD56 and CD57 (lower row) and had large granular lymphocyte morphology (not shown). MAb to TCR alpha/beta was positive and TCR gamma/delta negative. No expression of CD30 or CD1a was noted (left lower plot). |
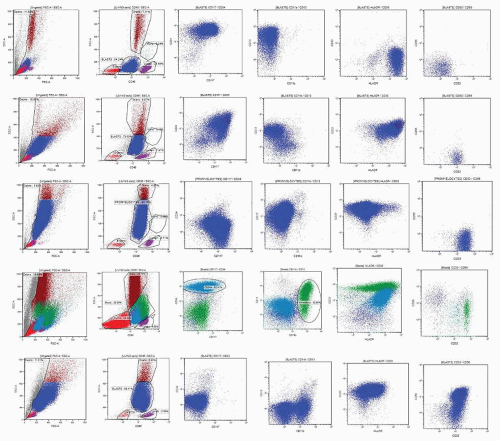 FIGURE 2.5. Examples of various scatter characteristics of CD45 dim blast population and patterns of antigen expression in acute myeloid leukemia. Bone marrow samples were stained with an eight-color MAb panel and acquired on a FACS-CANTOII flow cytometer (BD Bioscience). Panel consisted of CD56-FITC/CD13-PE/CD34 PerCP-Cy5.5/CD117-PE-Cy7/CD33-APC/CD11b APC-Cy7/HLA-DR Pac Blue/CD45 AmCyan. Analysis was performed using Kaluza software (Beckman Coulter). After removal of dead cells and debris, blasts, lymphocytes, monocytes, and granulopoietic precursors/granulocytes were gated on the CD45/SS plot. Further analysis of antigen expression was performed within the blast population (dark blue dots) except for myelomonocytic leukemia (fourth row) where the monocyte gate was added (green dots). The upper row of plots shows an example of AML without differentiation showing agranular blasts, positive for CD34, CD117, CD13, and HLA-DR, but negative for CD33 and CD56. The second row shows an example of AML with granulocytic differentiation as demonstrated by partial expression of CD11b and SS characteristics. Blasts are strongly positive for CD34, CD117, CD13, CD33, and HLA-DR but negative for CD56. The third row shows an example of APL with characteristic high SS and negative CD34, HLA-DR, CD11b, heterogeneous CD13, strong CD33, and no expression of CD56. The fourth row shows an example of myelomonocytic AML where a population of blasts (dark blue) and a population of aberrant monocytes were detected. Blasts were positive for CD34, CD33, CD11b, and HLA-DR but negative for CD117 and CD13. Both blasts and monocytes showed aberrant expression of CD56. The lower row shows an example of monoblastic leukemia, which was negative for CD34, CD117, and CD13 but showed strong expression of CD33 and CD56, dim HLA-DR, and partial expression of CD11b. |
by a decrease in CD44, disappearance of CD45 and acquisition of bright CD235a expression. At transition to the polychromatophilic/orthochromatophilic stage, erythroblasts show loss of HLA-DR, further decrease in CD44, and a mild decrease in CD36.51,52
 FIGURE 2.6. Flow cytometry analysis of maturation in granulopoiesis. Reactive bone marrow samples were stained with an eight-color MAb panel and acquired on a FACS-CANTOII flow cytometer (BD Bioscience). Panel consisted of CD56-FITC/CD13-PE/CD34 PerCP-Cy5.5/CD117-PE-Cy7/CD33-APC/CD11b APC-Cy7/HLA-DR Pac Blue/CD45 AmCyan. Analysis was performed using Kaluza software (Beckman Coulter). Granulopoietic cells and blasts were gated on CD45/SS plot within a live cell gate (upper left). CD34+ cells were gated in a live cell gate and a Boolean gate was created by adding both gates (called granulopoiesis). Expression of CD34 and CD117 showed three populations: CD34+/CD117-CD34+/CD117+ and CD117+/CD34-. The right upper plot shows maturation in granulopoiesis corresponding to promyelocytes (I: CD13+ CD11b-), myelocytes (II: CD13+/dim, CD11b dim), metamyelocytes/bands (III: CD13 dim, CD11bright), and mature neutrophils (IV: CD13bright, CD11b bright). The lower row of plots illustrates the position of these various subsets in other antigen expression plots. All granulopoietic cells were negative for CD56 (not shown). |
CD34+CD10+ Terminal deoxynucleotidyl transferase (TdT)+CD79a+CD19neg common lymphoid progenitor (CLP): early B (E-B) stage.
CD34+CD19+CD10+TdT+CD20-cytIgM- pro-B-cell stage.
After down-regulation of CD34 and TdT they become CD34-CD19+ CD10+ CD20 heterogenous pre-B that can be further subdivided in I and II subsets.
CD34-CD19+CD20+CD10dim/- IgM+ immature (IM)-B-cells.
After expression of light chains, cells become CD10-CD19+ CD20+ IgM+ IgD+ mature B-cells.
in reactive BM. Eosinophilic myelocytes can be identified by high side scatter, intermediate CD45 (at a level slightly higher than neutrophilic myelocytes), low to intermediate CD11b, intermediate CD13, and low CD33 with bright CD66b and no CD16 expression. Mature eosinophils show increased levels of CD45 and CD11b with a decrease in CD33 and are negative for CD16.51,64
TABLE 2.4 SURFACE MARKER EXPRESSION DURING MATURATION OF GRANULOPOIETIC PRECURSORS IN THE BONE MARROW | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 2.5 AVERAGE RELATIVE FREQUENCY OF MAJOR LYMPHOID CELL SUBSETS IN NORMAL TISSUES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


