B Lymphocytes
Frixos Paraskevas
ONTOGENY
Pluripotential stem cells give rise to all hematopoietic lineages, undergoing asymmetric division, i.e., one daughter cell follows a pathway of differentiation, whereas the other remains as a self-renewing stem cell. Depending on their interaction with the appropriate stromal cells, as well as growth and differentiation factors, stem cells differentiate into various lineages including B and T lymphocytes. Their differentiation is guided by the type of stromal elements to which they are exposed; i.e., in the thymic microenvironment they develop as T cells, whereas in contact with bone marrow-derived stromal cells they develop as B lymphocytes or myeloid cells. Multiple checkpoints ensure proper differentiation of precursor to mature B lymphocytes, and several have been found disrupted in patients with primary immunodeficiency. The differentiation of the B cell involves critical processes divided into two periods; the first from stem cell to immature Ig-M+ B cell is antigen-independent, while the second period from immature B cell to plasma cell is antigen-dependent. The first period is subdivided into two stages: from stem cell to progenitor B (pro-B) cell and from pro-B cell to immature Ig+ B cell. Differentiation has been studied in animals and humans with cells obtained from different stages of development (embryonic as well as adult) and with cells from patients with lymphoproliferative disorders, which are considered “frozen windows” of normal differentiation. This approach, however, is not accepted universally because evidence from acute leukemias suggests the existence of some degree of asynchronous maturation with phenotypes not found normally.1
STEM CELL TO PROGENITOR B CELL
See Figure 12.1
Transcriptional Regulation
Hematopoiesis is coordinated by several genes that orchestrate multiple cell interactions which release growth and differentiation factors.2 More than eight specific cell lineages have been identified which differentiate from pluripotent stem cells, guided through a well-defined hierarchical sequence to mature functional cells.3
During fetal life lymphopoiesis takes place in the yolk sac and liver and after birth in the bone marrow.4 “Master” genes regulate differentiation of lineages through transcription factors which activate target genes, progressively narrowing their differentiation potential to specific lineages,3 and which coordinate the various stages of lymphocytic development.5,6,7,8 A common progenitor gives rise to separate myeloid and lymphoid progenitors, and the latter to lymphocytic lineages B, T, natural killer (NK), and dendritic cells (DC).
Normal development of lymphopoiesis depends on the Ikaros family of transcription factors, which regulate lymphocytic development. The Ikaros gene (IKZF1) encodes a zinc finger protein (ZFP), which is a transcription factor expressed by B and T cells at different stages of differentiation (except plasma cells).9
A long-standing goal of molecular biology has been the construction of DNA-binding proteins for the control of gene expression by using the classical Cys2-His2 design of a ZFP, ideally suited for such purposes; and it was adopted for discriminating genes with closely related DNA sequences.10 The classical ZFs are one of the most common protein domains in higher eukaryotes widely used in zinc finger nucleases (ZFN) and are currently used in human clinical trials with an aim of therapeutic gene editing. ZFPs are nucleases designed to cut at specific DNA sequences and are becoming powerful tools in gene targeting to replace a gene within a genome by homologous recombination (HR), or to permanently modify plant and mammalian genomes, including the human genome. They were developed and used in Drosophila, rats, and zebra fish to generate modified organisms with genetargeted mutations and/or deletions.
This brief review emphasizes the point that Ikaros is a master regulator of gene expression and chromatin remodeling, and its biologic roles include regulation of the development and function of the immune system and overall even the hematopoietic differentiation.
The Ikaros-/- mice have a complete block of B-cell differentiation, with lack of pro-B and precursor B (pre-B) cells in fetal liver and bone marrow. The Ikaros gene generates eight protein isoforms by alternative splicing and some of them bind, through the N-terminal ZFP motif, to DNA sequences which contain GGGA core motifs. This family of DNA-binding proteins, restricted to lymphocytes, are highly conserved in humans and mice and regulate the expression of other genes. They are localized in the nucleus of most primitive hematopoietic stem cell subsets, particularly at two stages: (a) long-term self-renewing stem cells and (b) multipotent progenitors, which are not self-renewing, but able to differentiate into lymphoid-committed progenitors.11 The isoforms detected in hematopoietic stem cells differ from those detected in lymphoid progenitors, but all of them share two C-terminal zinc fingers that mediate their self-association, forming multiple heteromeric complexes. Other members of the Ikaros family (i.e., Aiolos and Helios) encode transcription factors that form multimeric complexes with Ikaros.12
The Ikaros gene is required not only for the early stages of lymphocytic differentiation but also for late stages, especially in T-cell maturation. It exerts multiple regulatory functions by recruiting repressor complexes, or what are known as chromatin remodeling machines, an unconventional function of transcription factors. IKAROS is also needed for the maintenance of B cells by regulating BCR signaling, and importantly overall it acts as a master regulator of hematopoietic differentiation and as a tumor suppressor in acute lymphoblastic leukemia (ALL), especially forms of the disease associated with poor prognosis.13
The transcription factor PU.1 acts at the pluripotent level of myeloid-lymphoid progenitors, and its expression maintains the hematopoietic progenitor pool by supporting the generation of the earliest lymphoid and myeloid progenitors.14
It regulates early B-cell development by activating the IL7Rα gene, rendering B-cell progenitors responsive to appropriate differentiation signals, and thus promoting differentiation to the pro-B-cell stage . Among the three main B-cell populations, B-1, B-2 (follicular), and marginal zone B cells, PU.1 directs differentiation toward the B-2 subpopulation15 and enhances the activity of other transcription factors, i.e., the interferon (IFN)-regulatory factor. The B-1 and B-2 B cells which are involved in innate and adaptive immune responses, respectively, arise from distinct progenitors.16
The TCF3 (previously E2A) gene is important for lymphocytic differentiation encoding two proteins, E12 and E47 of the basic helix-loop-helix family by differential splicing. The proteins encoded by TCF3 bind uniquely to the DNA of B cells as homodimers17 and are essential for the coordination of Ig gene
rearrangements;18 therefore in their absence B-cell differentiation is blocked before entrance into the pro-B-cell stage. The basic region mediates DNA binding, whereas the HLH domain is required for protein dimerization. The E12 targets the genes of the early B-cell factor (EBF)19 and the Pax5,20 directing differentiation along B lineage while it blocks myeloid differentiation.
rearrangements;18 therefore in their absence B-cell differentiation is blocked before entrance into the pro-B-cell stage. The basic region mediates DNA binding, whereas the HLH domain is required for protein dimerization. The E12 targets the genes of the early B-cell factor (EBF)19 and the Pax5,20 directing differentiation along B lineage while it blocks myeloid differentiation.
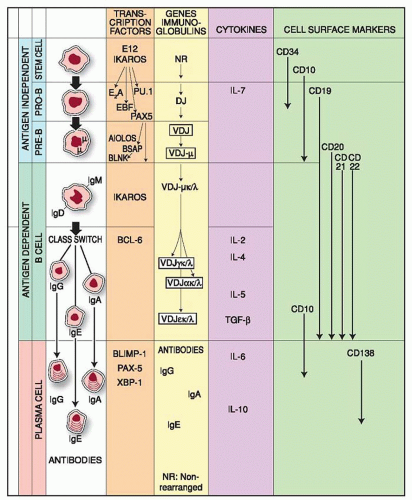 FIGURE 12.1. B-cell differentiation. The first stage of B-cell development takes place in the bone marrow. Normal development of lymphopoiesis depends on the Ikaros family of transcription factors, which form multimeric complexes with other members, i.e., Aiolos and Helios. These complexes are known as chromatin remodeling machines, and their targets induce a second wave of transcription factors, i.e., PU.1, E2A, EBF, PAX5, etc. Their targets are a third set of genes, forming BSAP that acts on the adaptor protein BLNK (SLP-65). Targets of BSAP are the genes for CD-19, V-preB, and λ5, i.e., markers distinctly of the B-cell lineage. BLNK regulates the transition from pro-B to pre-B, whereas PAX5 is needed for V(D) J recombination. This stage of development is independent of exposure to antigen and is followed by an antigen-dependent stage that unravels in the peripheral lymphoid organs. Antigen receptors, i.e., Igs, expressed on their surface interact with antigen and helper T cells, and trigger B-cell differentiation (plasma cells) and antibody secretion. Class switching generates distinct classes of antibodies suitable to dispose antigens in the various microenvironments, i.e., inside the body as well as on body surfaces. |
The Pax5 is exclusively expressed in the stage between pro-B cell and mature B cell.20 The B-cell-specific activator protein, BSAP/ PAX5, is a paired domain DNA-binding protein expressed in the developing nervous system, testis, and in all B-lineage cells, except terminally differentiated plasma cells. BSAP regulates transcription of several genes expressed in B cells and also the activity of the 3′-IgH enhancer and targets the genes of CD19, VPREB1, the gene transcribing for the lambda light chain, and several intronic sites of the C4B gene. The Pax5 gene regulates the V(D)J Ig heavy-chain gene rearrangements after the first DJ-H rearrangement,21 which generates the pro-B-cell population. Mice deficient for Pax5 have a pre-B-1 cell population which do not express CD19 or Ig-α and to a lesser extent Ig-β, V-preB, and lambda-5. These cells proliferate normally in vitro on stromal cells in the presence of interleukin IL7 and carry out D-J rearrangements of both IgH chain alleles. Removal of IL7 from the cultures inhibits differentiation to surface Igm expressing B cells. Transplantation of Pax5-deficient pre-B I clones into Rag-deficient hosts leads to full reconstitution of the thymus with D(H)J(H) rearrangements and normal positive and negative selection of CD4–CD8– and CD4+CD8+ thymocytes and the development of normal, mature CD4+ and CD8+ T-cell compartments in the peripheral lymphoid tissues.
The target for BSAP is the adaptor protein B-cell linker protein (BLNK) which is linked with surface IgM by CMTM7, a tetra-spanning membrane protein, co-localized with clathrin and sIgM on the plasma membrane. The linking of sIgM with BLNK, a pivotal adaptor protein in the signal transduction, results in its recruitment to the vicinity of the tyrosine kinase Syk and initiation of signal transduction. This culminates in MAP kinase activation as well as other downstream signaling proteins, i.e., PLC-γ2, Bruton tyrosine kinase ( Btk), etc.22 The BSAP regulation of BLNK gene expression controls also the pro-B to pre-B transition and mediates the constitutive signaling of the pre-B cell receptor (pre-BCR) in cell proliferation, growth factor responsiveness, and V(D)J recombination. BSAP diverts differentiation of a common myeloid/lymphoid progenitor to lymphocytic differentiation as a result of its ability to suppress the response of the progenitor cell to myeloid growth factors,23 and represses genes inappropriately expressed at the pro-B-cell stage.24
Essential for early B-lymphocyte development is the EBF which regulates the expression of Ig-α (mb-1) and Ig-β (B-29) coreceptors. Isolated human EBF factor binds to Ig-α and Ig-β promoters, and EBF-deficient mice lack Ig-α and Ig-β transcripts in pro-B cells.25,26 EBF activates transcription of B-cell-specific genes and initiates necessary events such as gene activation of other transcriptional regulators (i.e., Pax5) that direct the early stages of B-cell lymphopoiesis.27 Mutations of E2A or EBF arrest B-cell development at a stage earlier than that of PAX5, because recombinase expression is blocked and expression of pre-BCR, λ5, and V-preB genes is inhibited.27 The three transcription factors PAX5, TCF3 (E2A), and EBF form a cross-regulatory network with the TCF3 gene being the most potent B-cell regulator, essential for expression of PAX5 and EBF, which in turn is needed for expression of E47. Collectively, the targets of IKZF1 are the genes of the transcription factors TCF3 (E2A), PU.1, EBF, and PAX5, all of which regulate stages of B-cell differentiation before the expression of Ig genes. It has been shown however, that the TCF3 gene plays a central role for some aspects in subsequent stages of B-cell lymphopoiesis.28 TCF3 (E2A) proteins are required for the interleukin IL7-dependent expansion of pro-B cells and their progression to the pre-B-cell stage and even later, for the regulation of rearrangements of the IgK gene and heavy-chain isotype switching.29,30,31 TCF3 encodes proteins that enhance hypermutation by recruiting AID to the Ig loci29 and is essential in promoting pre-B- and B-cell survival.30
CELL INTERACTIONS IN EARLY B-CELL DEVELOPMENT
Study of B-cell differentiation has become feasible with the development of long-term bone marrow culture techniques. This approach has helped to define the cells that are essential for the development of B cells and the factors that support B-cell growth and differentiation. The bone marrow stroma makes a critical contribution to hematopoietic differentiation and the term stroma is used in a collective sense to include a variety of cells, such as adventitial reticular cells, adipocytes, fibroblasts, and endothelial cells of the sinuses.32 The stroma includes also the extracellular matrix, composed of various fibrous proteins, glycoproteins, and heparan sulfate, effective in binding cytokines.
Granulocyte-macrophage colony-stimulating factor can bind to marrow stromal glycosaminoglycans and be presented to hematopoietic stem cells. Stroma cells have been isolated from adult bone marrow33 or fetal bone marrow after removal of adherent cells (i.e., macrophages, endothelial cells). Stroma cells have the morphology of an adventitial/reticular/fibroblast cell, which expresses several adhesion molecules. They support, in cultures, differentiation of fetal bone marrow cells (CD34+, CD19–)34 toward B-cell lineage, i.e., with loss of CD34 and acquisition of CD19 and the V-pre-B protein.
Several adhesion molecules have been implicated in the mediation of interactions between B-cell progenitors and stroma cells. Critical among them are vascular cell adhesion molecule-1 (VCAM1) on stroma cells and very late antigen-4 (VLA-4) on B-cell progenitors.33,35 The CD34 is expressed not only on stem cells,36 but also on stroma cell precursors37 and endothelial cells,38 and thus mediates interactions with its counterreceptor, L-selectin expressed on progenitor cells. Among cytokines with a wide range of functions is IL7, which acts in an early stage of common lymphoid progenitors (CLP, B/T/NK), mediates growth and differentiation of lymphocytes in the bone marrow, and plays a critical role in B-cell development.39 IL7 maintains B-cell differentiation programs open; and although in the absence of IL7 the differentiation of T/NK proceeds normally, B-cell differentiation is arrested since IL7 modulates EBF expression, which activates target genes that are specific for B-cell differentiation programs.40
In mice lacking IL-7R the recombination process comes to a halt, because normally it is the IL-7R which targets VH segments to the recombinase complex; and furthermore, aberration in the expression of Pax5, a gene with pivotal importance in B-cell development and regulation, is associated with B-cell malignancies.41 Even though the block from IL7 deficiency is after the pro-B cell stage, these cells have certain abnormalities, such as failure to upregulate terminal deoxynucleotidyl transferase (TdT) and the high-affinity IL-7R chain.42 The net result of IL7 deficiency is lack of expression of the µ-chain and the pre-BCR, events that normally promote the advancement from pro-B-cell to pre-B-cell stage. Stimulation of proliferation and expression of the µ-chain follows distinct signaling pathways, because the Y449F mutation in the cytoplasmic region of the l7rα chain, which abrogates proliferation induced by IL7, does not block µ-chain expression.43 In the absence of IL7, the thymic stroma lymphopoietin (TSLP) (see Chapter 11) assumes a host of functions, i.e., regulation of the development of IgM+ B cells from IgM– precursors, the promotion of B-cell and Th-2 differentiation of naive CD4+ T cells, and the activation of NK T cells, basophils, and other innate immune cells at the initial stage of inflammation.
TSLP affects B cell maturation and activation and may also influence differentiation and development of T regulatory T (Treg) cells.44 Human X-linked severe immunodeficiency results from mutations of the γ chain, a common subunit for several cytokine receptors (i.e., IL2, IL4, IL7, IL9, and IL15). The patients lack T and NK cells but have normal or even elevated B cells,45 and normal numbers of B cells are detected in patients with mutations of the IL7Rα chain.46 The IL7 exerts changes in gene expression, which effect differentiation of pro-B cells to pre-B cells, especially in concert with other growth factors of stromal cells.47 It synergizes with FMS-like tyrosine kinase 3 ligand (FLT3LG) and induces strong expansion of fetal B cells in vitro.48 IL7R has been detected on human B-cell progenitors, with a pro-B-cell phenotype, which lack expression of CD19 and clonogenic capacity,49 are CD34+, have messenger RNAs (mRNAs) for CD79B, RAG1, PAX5, and are TdT+. The IL7R-α–/CD19–/CD34+ cells do not differentiate in short-term cultures into pro-B cells, therefore the expression of IL7R-α defines an entry into a stage characterized by upregulation of multiple B-lymphoid-associated markers. Single adult human CD34+ CD38– CD7– cells cultured over a murine fetal liver adherent cell line supplied with IL7, FLT3LG, and IL3 differentiate into B cells, NK cells, myeloid cells, and DC.50
B-lineage development therefore is based on a complex, interconnected network of genetic programs which provide lineage commitment and specification.8,51
The CXCR4 chemokine receptor is detected in CD34+ hematopoietic progenitors52 committed to B-cell development,53 and its ligand is the stromal cell-derived factor-1 (SDF1 or PBSF, now known as CXCL12).54 The CXCL12 chemokine orchestrates rapid revascularization of injured, ischaemic, and regenerating tissues via the CXC chemokine receptors CXCR4 and CXCR7. The CXCL12 induces intracellular actin polymerization in lymphocytes, a process that is thought to be a prerequisite for cell motility. Expression of CXCR4 on CD34+ BM cells is one of the phenotypic alterations for committed lymphoid progenitors and allows discrimination of lymphoid progenitors from myeloid progenitors. Since CD34+CXCR4+ cells are differentiated from CD34+CXCR4– cells, multipotential progenitors located in the BM are likely to be negative for CXCR4 expression.
CXCR4 is also known as fusin and acts as a coreceptor for human immunodeficiency virus (HIV) which infects host cells, but CXCL12 blocks the entry of HIV to T cells.53 The CD34+ CXCR4– bone marrow cells generate myeloid and lymphoid progenitors, while the CD34+, CXCR4+ cells, although they give rise to B cells and T cells, lack the potential for myeloid, erythroid, megakaryocytic, and mixed colony-forming progenitors. CXCR4 therefore defines B-lineage committed progenitors more accurately than other commonly used markers and is expressed earlier than IL7R or TdT.
PRO-B CELL TO IG+ B CELL
The early development of B cell differentiation is divided into two stages, the pro-B and the pre-B cells. In pro-B cells the first rearrangement involves the DH to JH segments of the immunoglobulin heavy chain (IGH@). With the completion of this step, the VH to DH-JH rearrangement begins and if it is productive, an H chain is formed which binds a surrogate light chain (SLC) forming the pre-BCR. Further signaling by the pre-BCR stimulates proliferation followed by light chain rearrangement. IGK@ light chain rearrangement begins before the rearrangement of the IGL@ light-chain genes. These epigenetic changes regulate the transition to the B cell stage, characterized by rearrangements of the light-chain genes with the kappa light chain preceding the rearrangement of the lambda chain gene.
Molecular definition of the stages of early B-cell development identified cells with µH chains in the cytoplasm without L chains and no mature IgM molecules on the cell surface,55 followed by phenotypic markers or Ig gene rearrangement.56 Single-cell polymerase chain reaction analysis and Ig gene rearrangements allowed a more precise identification of B-lineage commitment and definition of the stages which characterize the transition from stem cells to pro-B-cell stage and B-cell development (Fig. 12.1). The pro-B cell stage in B cell development is followed by the pre-B cell, which may be divided into pre-B-I and pre-B-II stages.
Clones of pre-B I cells carry characteristic D(H)J(H) rearrangements on both IGH@ chain alleles. However removal of IL7 from the tissue cultures does not induce B-cell differentiation in B cells expressing surface IgM.
When the Ig gene rearrangements reach their final stage, IgM is expressed on the cell surface and differentiation reaches the end of the immature B lymphocyte stage. Three sets of genes regulate the transition from the pro-B-cell to the pre-B-cell stage, with expression of the complete Ig molecule on the surface of the B cell. One set includes RAG1 and RAG2 genes, and a third gene encodes the SLC which is composed of the proteins from the VPREB1 and λ5 genes.
The proteins from two other genes, CD79A (mb1, PSMBS; IGB) and CD79B (CD79B; IGB) CD79B; IGB, form the coreceptor. These
genes support the expression of IgM on the surface of the B cell, the BCR repertoire, and allelic exclusion (i.e., expression of only one of the two alleles). The rearrangements of the Ig genes start with the joining of a D to JH segment in the pro-B cells, which proliferate in contact with stromal elements and express the SLC linked to a gp30 protein, sometimes referred to as the surrogate heavy chain of the pro-B-cell complex.57,58 and 59 SLC expression is required to establish immunoglobulin heavy chain allelic exclusion during early B-cell development and is easily detected on mouse pro-B cells but difficult to detect on human pro-B cells.58,59 The SLC consists of two noncovalently associated proteins, VPREB1 and λ5 (Fig. 12.2), encoded by genes located on chromosome 22, where the λ-chain genes are also located.60,61 The VPREB1 and λ5 proteins have several unusual features that distinguish them from conventional light chains. The VPREB1 protein is composed of 102 amino acids with homology to a conventional V domain and both have atypical sequences which target them to the endoplasmic reticulum (ER).62
genes support the expression of IgM on the surface of the B cell, the BCR repertoire, and allelic exclusion (i.e., expression of only one of the two alleles). The rearrangements of the Ig genes start with the joining of a D to JH segment in the pro-B cells, which proliferate in contact with stromal elements and express the SLC linked to a gp30 protein, sometimes referred to as the surrogate heavy chain of the pro-B-cell complex.57,58 and 59 SLC expression is required to establish immunoglobulin heavy chain allelic exclusion during early B-cell development and is easily detected on mouse pro-B cells but difficult to detect on human pro-B cells.58,59 The SLC consists of two noncovalently associated proteins, VPREB1 and λ5 (Fig. 12.2), encoded by genes located on chromosome 22, where the λ-chain genes are also located.60,61 The VPREB1 and λ5 proteins have several unusual features that distinguish them from conventional light chains. The VPREB1 protein is composed of 102 amino acids with homology to a conventional V domain and both have atypical sequences which target them to the endoplasmic reticulum (ER).62
However, whereas the conventional V domain has a total of nine beta strands, including a sequence encoded by a J segment, the VPREB1 has no sequence similar to the J segment and is shorter by one strand, the β7 strand, and its C-terminal portion has no homology to any other known protein. The N-terminal end of the λ5 protein is unique, with only marginal sequence homologies to conventional domains. The C-terminal end of the λ5 protein has an Ig-like fold, homologous to the conventional λ-chain C domain. This is followed by a sequence with marginal homology to the Ig structures and an extra β-strand that is homologous to the J region of the conventional Vλ domains. Unique regions and Ig-like domains of the VPREB1 and lambda-5 proteins control the folding and assembly of SLC,62 which associates with the µ-chain to form the pre-BCR. The C-1 domain of the µ-H chain in the pre-BCR is protected from improper folding by the hsp-70 protein BiP, which retains the µ-H chain in the ER.63 The protection from improper folding of CH-1 is taken over by the C-λ5 protein at a later stage as long as the CH-1 is associated with the VPREB1 as SLC.64 SLC escorts the µ-H chain to the surface of the cell, although not all µ-H chains can pair with the SLC, since some V segments have structural features that prevent their association. At this point, the RAG1, RAG2, and DNTT (TDT)TdT genes are turned off, securing allelic exclusion and thus preventing further rearrangements at the other allele.65 The λ-5 chain of the pre-BCR represents the C domain of the conventional L chain of BCR, and has 85% homology with the C-λ domain, while the VPREB1 is the equivalent to the VL domain.66 The SLC is linked by a disulfide bond to the µH chain through the Cµ-1, i.e., the first constant domain,67 and its role in the formation of the pre-BCR is dual. The C domain of λ-5 protein interacts with Cµ-1 and the VPREB1 (and part of λ5) with the VH domain of the µH chain.68 This last interaction is likely to be the first step in the selection of the B-cell repertoire67 as the pre-BCR induces asymmetric divisions of pre-B cells.69 In contact with pre-BCR ligands on stromal cells, i.e., heparin sulfate70 or galectin-1,71 the SLC mediates receptor aggregation, signaling, and internalization, as well as transition from pro-B cells to pre-B cells.72 Pre-B-cell signals induce proliferative activity of pre-B cells, allelic exclusion of the heavy-chain locus, and activation of the light-chain loci preparing for V(D)J recombination.73
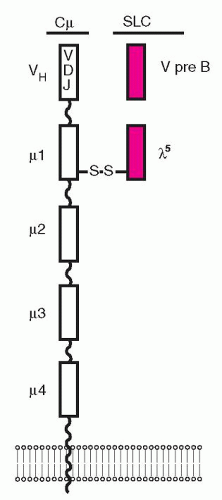 FIGURE 12.2. The structure of the pre-B-cell receptor. The pre-B-cell receptor is composed of the µ chain and a surrogate light chain that consists of two components, the λ5 and the V-preB. The µ chain is linked by disulfide bond to the λ5 component, whereas the V-preB is noncovalently attached. It is not yet clear whether the surrogate light chain can be expressed on the surface without the µ chain. |
Productive rearrangement of the L chain completes induction of the IgM molecule expressed on the B cell surface, and marks the transition to the immature B-cell stage. The strong proliferative capacity of the pre-B cells was analyzed in Pax5-/- mice, which showed that the pre-B cell has an extensive renewal capacity, multipotency, and the ability to reconstitute recipient animals with lymphoid and myeloid lineages.74
Signaling is transmitted by the CD79A/CD79B partners of the pre-BCR, which is linked to several protein tyrosine kinases, i.e., Bruton’s tyrosine kinase [BTK], SYK, ITK, BLNK, and phosphoinositide 3-kinase (PI-3K).75 Ligand binding causes aggregation of the pre-BCR, resulting in endocytosis of pre-BCR, loss of cell surface expression, and accumulation of pre-BCR within membrane lipid microdomains.
The pre-BCR signaling achieves two important outcomes for subsequent differentiation of the B cell: activation of MAP kinases, which block apoptosis usually triggered by the engagement of CD24;76 and downregulation of RAG1/RAG2 expression to turn V(D)J recombination off, so that allelic exclusion is accomplished.77 The RAG1/RAG2 proteins are still expressed on active proliferating cells of the pre-BI stage, but are silenced during the pre-BII stage. Because the cell has achieved its objectives (i.e., survival and allelic exclusion), it reactivates the V(D)J recombination machinery to allow rearrangements of the conventional κ/λ chains, to be able to form the complete IgM molecule and thus enter the immature B-cell stage.
Existing epigenetic mechanisms prioritize expression of single heavy and light chain alleles (allelic exclusion) in order to maintain monoallelic expression of both genes in a single B cell,78 maintaining at the same time single specificity of the B cell receptor. At a later stage of B cell development, additional mechanisms contribute to prioritizing expression of single IGH@ and IGL@ alleles to help maintain the monoallelic expression of Ig.
The nontranscribed allele is in close association with heterochromatin, and the transcription factor IKZF1 initiates the silencing of transcription of the lambda-5 gene through a direct effect on the promoter.79 Individual B lymphocytes normally express immunoglobulin (Ig) proteins derived from single Ig heavy chain (H) and light chain (L) alleles. At later stages of B cell development, additional mechanisms may contribute in prioritizing expression of single IGH@ and IGL@ alleles. The importance of pre-BCR signaling for progression from pro-B-cell to pre-B-cell stages and beyond is emphasized by the fact that deficiencies of any component of the pre-BCR or downstream signaling molecules blocks developmental progression of B cells at the pro-B or pre-B stage.80 Targeting the genes for VPREB1,81 λ5,82 or exons of the µ-chain blocks B-cell differentiation at the pro-B to pre-B-cell stage.83 In mice with λ5-deficiency the expression of VPREB1 is normal, which excludes an essential role for pro-B and pre-B
cell receptors in VPREB1 regulation.84 Similarly, in a patient with agammaglobulinemia, mutations in both λ5/IGLL1 genes were associated with agammaglobulinemia and reduced numbers of B cells.85
cell receptors in VPREB1 regulation.84 Similarly, in a patient with agammaglobulinemia, mutations in both λ5/IGLL1 genes were associated with agammaglobulinemia and reduced numbers of B cells.85
The SLC is not detected on the surface or in the cytoplasm of a subpopulation of self-reactive cells found in germinal centers86 and circulation.87 B cells having a VPREB1 sequence in their BCR and RAG mRNA have also been described in normal human tonsils and the joints of patients with rheumatoid arthritis.88,89 The pre-B cell signaling that terminates pre-B cell expansion and induces Ig L-chain rearrangement is transmitted by Bruton’s tyrosine kinase and the BLNK (Src homology-2 domain), which contains a leukocyte-specific phosphoprotein of 65 kD.90 The V-region of a pre-B receptor and λ5 proteins of the pre-BCR are invariant, but each contains unique evolutionarily conserved non-Ig sequences (unique regions or URs) attached to the Ig sequences.91 The URs interrupt the SLC complex and may be considered equivalent to the CDR3 of the Ig L chain which participates in antigen binding.92 The transcription factors that are most active and are specifically upregulated at this stage are TCF3, EBF1, PAX5, and IRF4.93 The pre-BCR not only switches on the Ig genes, it appears to select preferentially those that provide µH chains of a higher quality, i.e., with a higher potential to assemble with Ig L chains.94
In conclusion, the pre-BCR plays a major role in the expansion of pre-B cells, allelic exclusion, repertoire selection, activation of V(D)J recombination, and developmental progression to IgM+ B cells.73 Multiple checkpoints ensure proper differentiation of precursor and mature B lymphocytes. Many of these checkpoints have been found disrupted in patients with primary immunodeficiency; and it still remains unclear how B cells are affected, as 11 critical processes in B cell differentiation have been identified.
IMMUNOGLOBULIN GENES
Among the early successes of investigations in recombinant DNA, was the characterization of the Ig genes. There are three loci for the heavy chains and two for the light chains (κ, λ). The gene for the human heavy chain is located on chromosome-14,95 the C(κ) gene on chromosome-2,96 and the C-λ on chromosome-22.97,98 Separate genes encode the V and C domains of the Ig molecule. The human heavy-chain genes are not completely linked and the Cµ gene is the closest to the Vdj segments.
At the 3′ end of the H chain locus, there are two copies of a γ–γ–ε–α unit (Fig. 12.3). One of the duplicated ε-sequences is a pseudogene (ψε) in which the CH1 and CH2 domains have been deleted. The genome contains a third closely homologous ε-related sequence, a “processed” pseudogene found on chromosome-9. These kinds of pseudogenes probably are products of a reverse transcription from RNA and then are inserted at locations in the genome that are unrelated to the original locus. A γ-related pseudogene lacking the switch region is present between the two γ–ε–α duplications. The recombination events occur in a region on the 5′ side of the CH coding sequences.
This region contains repetitive DNA sequences and has been known as the switch region (S) sequence. Within the switch regions occur the recombination breakpoints during isotype switch (see Chapter 14). Usually the stimuli for switch recombination, like IL4 and CD40, promote transcription across the S regions to produce switch junctions, one retained in the chromosome while the other is found in a circular DNA (see Chapter 14).
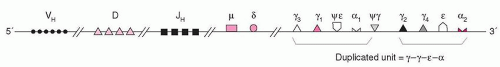 FIGURE 12.3. Human immunoglobulin heavy-chain gene locus. The heavy-chain gene locus contains the µ and δ genes close to the JH genes, whereas the remaining heavy-chain genes are farther downstream in two clusters from duplications. See text. |
VH-D-JH GENES
The heavy chain of the IgM molecule is encoded by four sets of DNA: variable (VH), diversity (D), joining (JH), and the constant (C-µ) genes.
A VH gene encodes the first 95 or 96 amino acids of a V domain but the exact number of VH genes is not known; it is estimated, however, to be 100 to 200. Of the available VH genes only a portion are functional, and an even smaller portion is available for rearrangement.99,100 The VH genes constitute seven families based on their homology at the DNA level and are known as VHI to VHVII. Most VH genes are polymorphic, but the degree of variation is usually small.101 The VH segment encodes the hydrophobic leader sequence, the three framework regions, and the two complementarity-determining regions (CDR).
The name D gene, or segment, was proposed to indicate the diversity of an antibody at positions after amino acid 99 to the beginning of the J segment,102 which spans the third CDR. The D segments are located in chromosome band 14q32, with the major locus between VH and JH loci and several D segments interspersed within the VH segments, sandwiched between signal sequences (see later).The CDR-3 is encoded by the 3′ end of the VH segment, the D gene segment, and the D-JH junctional area. There are approximately 30 D segments grouped in seven families, and each one spans approximately 70 kb, with a promoter on its 5′ end that allows initiation of transcription when Ig genes at the pre-B-cell stage start rearrangements. The first rearrangement produces a DJH complex, which together with the µ gene, encodes a protein known as Dµ protein. There are six functional JH genes and three pseudogenes, each JH gene encoding the 3′ end of CDR-3 and the fourth framework region.
V (D) J RECOMBINATION: FORMATION OF THE CODING AND SIGNAL JOINTS
Next to the V, D, and J genes are conserved sequences of seven nucleotides (heptamers), which are attached to the 3′ side of the V segments, the 5′ side of the J segments, and on both sides of the D segments (Fig. 12.4). The heptamer is followed by 12 or 23 nonconserved base pairs (bp) (spacers), followed by another sequence of 9 bp (nonamers), which may diverge from the consensus sequence. This noncoding sequence is known as the recombination signal sequence (RSS).
Recombination signal sequences for V(D)J joining consist of a conserved heptamer (CACAGTG) and a nonamer (ACAAAAACC) separated by a spacer of a constant length of 12 or 23 bp. The presence of three consecutive A residues in the nonamer is necessary for efficient recombination, and the nucleotides flanking the A-rich core should be other than A residues, so that the border of the A-stretch is precisely defined. This may be important when the recombinase measures the distance between the heptamer and the nonamer to satisfy the 12/23-bp spacer rule.103
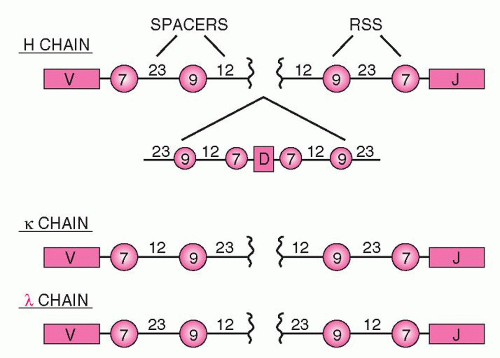 FIGURE 12.4. Recombination signal sequences (RSS) and heptamer/nonamer spacing sequences. The coding sequences of the immunoglobulin genes are flanked by a 7-nucleotide sequence (heptamer), which is followed by either 12 or 23 nucleotides, which in turn are followed by a 9-nucleotide sequence (nonamer) and again by a 23- or 12-nucleotide spacer. This order allows rearrangements only between a segment in which the 7/9 sequences are separated by a 12-mer spacer and another segment in which they are separated by a 23-mer spacer. This is known as the 12/23 rule. |
The 12-bp spacer corresponds to one turn of the DNA α-helix, whereas the 23-bp spacer corresponds to two turns. This way, the recombining segments are juxtaposed on the same side of the DNA helix, so that they can be recognized by the enzymes of the recombination machinery.104,105 Joining of the various segments is limited between an RSS with a 12-bp spacer and one with 23 bp106 (the 12/23 rule). Recombination follows strictly the 12/23 rule of spacers, which prevents inappropriate recombination, i.e., a VH segment joining directly to a JH segment, as both of them have a 23-bp spacer between their heptamers and nonamers.106 The pattern of the RSS at each locus is uniform. For example, in the Ig locus, all Vk segments have next to their heptamers a 12-bp spacer, whereas all Jk segments have a 23-bp spacer. This prevents accidental joining of two Vk or Jk segments. The RSS sequences are the only ones that are required for recombination, whereas the coding sequences (V, D, or J) can be replaced by other DNA while the joining still occurs if the 12/23 rule is satisfied. During recombination, two new structures are formed: the joining of the coding sequences (coding joint), which is imprecise, and the joining of the RSS (signal joint), which is precise.
The V(D)J recombination (Fig. 12.5) is mediated by two enzymes known as RAG proteins,107 the RAG1 and RAG2, which are not related to other proteins. Their expression normally is limited to immature B and T cells, even after the expression of surface antigen receptor, until the receptor is cross-linked. Deficiency of RAG proteins in mice108,109 or humans110 results in SCID. The RAG2 is divided into an N-terminal (core) domain and a C-terminal (non-core) domain. The core domain is necessary and sufficient for carrying out the V(D)J recombination. The C terminus of RAG2 contains a PHD finger motif, usually present in chromatin-associated proteins. The PHD (Plant Homeodomain) is encoded by many genes, including RAG2, and recognizes or “reads” the gene status of expression for histone H-3, lysine-4, and DNA damage, i.e., histone modifications, and as a result is known as a chromatin reader. The PHD finger of RAG2 modulates V(D)J recombination,111 and mutations of RAG2 at this site result in immunodeficiencies. Tetramers of the RAG1/RAG2 proteins initiate the V(D)J recombination, which proceeds in three steps.112,113,114,115 The RAG1 binds to the nonamer, which acts as an anchoring platform, while the heptamer stabilizes the complex in the presence of RAG2. The recognition by RAG proteins of the RSS is assisted by sequence-nonspecific DNA-binding proteins, HMG1/2 (high-motility group), which enhance binding and cleavage. In the RAG1/RAG2 complex, the RAG2 C terminus probably interacts directly with histones and stabilizes the RAG1/RAG2 complex.116 This interaction, which requires Mg2+, may bring the recombinase to specific signal sequences which may be required for the postcleavage phase of the recombination. HMGB proteins bind to DNA through two DNA-binding domains known as HMG-box-A and -B.117 Each consists of 80 amino acids interacting with the minor groove of the DNA. Box-A bends distorted DNA structures, whereas box-B bends severely linear DNA sequences. Mutations in HMGB proteins block synaptic complex formation, or possibly recognize distorted DNA structures induced initially by RAG1/RAG2 binding. Their binding stabilizes the formation of the synaptic complex and thus promotes the DNA cleavage.118 The recognition by RAG proteins of the RSS is assisted by sequence-nonspecific DNA-binding proteins, HMG ½, which enhance binding and cleavage. During the first step of V(D)J recombination, the RSSs are recognized by the recombinase and are brought in juxtaposition, forming the synaptic complex, which is composed of a dimer of RAG-2 and at least a trimer of RAG1.119 In the second step the recombinase nicks the phosphoester bond between the last nucleotide of the coding sequence and the first nucleotide of the RSS (Fig. 12.5, Panel 1). The precise mechanism of the hydrolysis of the phosphate ester bond is not known. The nicking reaction requires the physiologic divalent cation Mg2+. The cleavage leaves blunt phosphorylated signal ends and hairpin sealed coding ends (Fig. 12.5, Panels 2 and 3), which are formed between the 3′-OH of the coding sequence (top strand) and the central phosphorus atom of the phosphate group between the coding sequence and the RSS of the opposite strand (the lower strand in Fig. 12.5). Formation of the hairpin requires significant bending of one or both DNA strands (Fig. 12.5, Panel 4). In the third step, the hairpin must be nicked open, so that the two coding ends form the coding joint. Opening of the hairpin may be done by a DNA repair complex120 or by RAG proteins themselves.121 Because the nicking may not be exactly in the center of the hairpin, the opening creates an overhang in one strand formed by the nucleotides from the other strand (Fig. 12.5, Panels 5 and 6). These are known as P-nucleotides, which are coming from the opposite or complementary end of the hairpin or palindrome (from the Greek palindromicos, meaning successive movement forward and backward, like the piston of an engine). As a result, they have an inverse complementary relationship with the adjacent coding end. Most of the coding ends detected in normal lymphoid precursors have 3′ overhangs.122 TdT adds nucleotides known as N-nucleotides (nongermline) to the strand with the missing nucleotides (Fig. 12.5, Panels 7 and 8). Disruption of TdT drastically lowers junctional insertions. In fetal and neonatal periods, the N insertions are low or absent as a result of the developmental regulation of TdT. The alterations in the coding joint as a result of P- and N-nucleotides are the basis for junctional diversity (Fig. 12.5, Panel 9).
The RAG proteins continue to be bound to the signal and coding ends in a four-end complex, known as a cleaved signal complex, at least while the coding ends are still processed. The RSSs are highly conserved among vertebrates, from sharks to humans. The three heptamer nucleotides closest to the recombination site are the most important, whereas mutations at other heptamer positions still allow recombination. Variations in the position of RSS may influence the use of gene segments IGK@ vs. IGL@.
The RSS joint is assumed to be lost by being converted to reactive, broken DNA, disposed of in the next cell division (deletional recombination). However, the existence of inverted segments of DNA in these loci indicates that sometimes signal joints are retained in the chromosome (invertional recombination), a mechanism that restores chromosomal integrity. The fate of the signal joint has taken an unexpected direction as a result of findings indicating that the RAG proteins perform genetic transposition
(i.e., act as transposases).123,124 The name given to the enzyme indicates its function, i.e., transposing or inserting pieces of DNA in a new location. The transposase MuA of the bacteriophage Mu is the best characterized in this class of enzymes, consisting of a tetramer in which two of the subunits nick the two ends of the Mu genome, while the other two catalyze the transfer of the ends into the target DNA. These reactions are important in the transmission of drug resistance among bacteria and the integration of retroviruses, such as HIV and others, into the genome. In fact the active site of MuA exhibits a striking similarity to the HIV integrase. It is interesting that the RAG1/2 act as tetramers in V(D)J recombination and resemble the HIV integrase responsible for inserting DNA copies of the viral genome into the cellular chromosomes. There are also similarities between the mechanisms of transposition and V(D)J recombination, suggesting that the RAG proteins may be members of the retroviral integrase superfamily.125 Hairpins during V(D)J recombination are the targets of the RAG-mediated transposition. Notable differences, however, exist in the nick-cleavage mechanisms between the two.126 Transposition of RSS in various positions, targeted to DNA, results in branched molecules.127,128 The high Mg2+ concentration prevailing in mammals, however, results in removal of transposed DNA by disintegration. No transposition in vivo has been reported as yet, probably because it is suppressed by GTP
and the C terminus of the RAG2 protein.129,130 Active transposition events are detrimental to the host because of the mutagenic potential of genomic rearrangements.
(i.e., act as transposases).123,124 The name given to the enzyme indicates its function, i.e., transposing or inserting pieces of DNA in a new location. The transposase MuA of the bacteriophage Mu is the best characterized in this class of enzymes, consisting of a tetramer in which two of the subunits nick the two ends of the Mu genome, while the other two catalyze the transfer of the ends into the target DNA. These reactions are important in the transmission of drug resistance among bacteria and the integration of retroviruses, such as HIV and others, into the genome. In fact the active site of MuA exhibits a striking similarity to the HIV integrase. It is interesting that the RAG1/2 act as tetramers in V(D)J recombination and resemble the HIV integrase responsible for inserting DNA copies of the viral genome into the cellular chromosomes. There are also similarities between the mechanisms of transposition and V(D)J recombination, suggesting that the RAG proteins may be members of the retroviral integrase superfamily.125 Hairpins during V(D)J recombination are the targets of the RAG-mediated transposition. Notable differences, however, exist in the nick-cleavage mechanisms between the two.126 Transposition of RSS in various positions, targeted to DNA, results in branched molecules.127,128 The high Mg2+ concentration prevailing in mammals, however, results in removal of transposed DNA by disintegration. No transposition in vivo has been reported as yet, probably because it is suppressed by GTP
and the C terminus of the RAG2 protein.129,130 Active transposition events are detrimental to the host because of the mutagenic potential of genomic rearrangements.
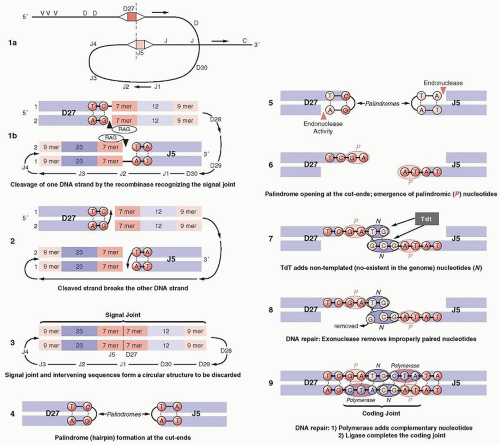 FIGURE 12.5. V(D)J recombination. Shown in this figure is the recombination of two hypothetical segments, D27 and J5. The recombination begins with recombination-activating gene-1 (RAG-1) and RAG-2 recognizing the heptamers and monomers (see text) (Panel 1). They cut out one strand at the junction between the heptamer and the coding sequence. The other strand is severed by a nucleophile attack from the cut end (Panel 2). The two blunt ends of the heptamers form the signal joint (Panel 3), while a hairpin seals the cut coding ends (Panel 4). The hairpin is nicked open either by the RAG proteins or by a ubiquitous DNA double-strand repair complex (Panels 5 and 6). The opening is usually asymmetric and creates overhangs, with the nucleotides at the end of the overhang being complementary because they derive from the complementary opposite strand. These are known as palindromic (P) nucleotides (Panels 5 and 6) (from the Greek palindromicos, meaning moving forward and backward in succession). The double DNA strand breaks of the coding joint receive nucleotides from the function of TdT (nongermline nucleotides) known as N-nucleotides (Panel 7). Improperly paired nucleotides are removed (Panel 8), and the coding joint is completed with the addition of missing nucleotides (Panel 9). The P- and N-nucleotides added to the coding joint constitute the junctional diversity. |
As a result of these recent insights into the nature of V(D)J recombination and the function of RAG1/2, the question of the fate of the signal joint becomes very important. Signal joints have been found to be cleaved quite efficiently.128 In vitro experiments showed that transposition is targeted not to a particular DNA sequence, but to structural features, such as hairpins, inverted repeats, supercoiling of DNA forming cruciform structures, and generally distorted DNA structures.131,132 In vitro experiments demonstrating transposition were performed with only the core part of RAG1/2; it is conceivable that other parts of the molecule exert an inhibitory effect on this potential function of RAG1/2. Although the in vitro experimental evidence suggests that there is a bias toward transposition, there is no evidence that this occurs in vivo, as it severely compromises genomic stability in the lymphocytes. On the other hand, lymphoid malignancies are associated with chromosomal translocations, many of which involve the Ig or T-cell receptor (TCR) loci and potentially may be mediated by the V(D)J recombinase.133
DNA REPAIR MECHANISMS
Cleavage of DNA is always potentially dangerous, and although the recombinase function is essential for the integrity and normal function of the immune system, it is, at the same time, perilous. The V(D)J recombination, as well as class switch recombination, are necessary processes, but they demand double-strand DNA breakage. As vital as these two processes are for survival, the DNA double-strand break (DSB) is a threat to survival and it does occur in viral infections and malignancies. If genetic integrity is to be maintained, the DNA breaks need to be repaired.134 Although nature has developed a highly complex repair mechanism, it is, however, efficient. Double-strand breaks generate an alarm mechanism, based on the transduction of signals from the sensors to the transducers and eventually to the effectors for the DNA repair job. An important sensor is the MRN complex, consisting of a nuclease (Mre-11) and the chromosome protein (RAD50), which maintain chromosomal structure and protein NBN.135,136,137 RAD50 and NBN are recruited rapidly to DSB sites for processing broken ends. MRN transduces the early signals to a kinase, the main transducer, known as ATM (ataxia telangiectasia mutated), which rapidly phosphorylates various substrates needed in the repair pathway. ATM belongs to a family of proteins known as PI-3-K-like protein kinases (PIKK), with five members, while an additional protein is the catalytic subunit of the DNA-PK kinase. Four of the PIKKs are involved in the DNA damage response: DNA-PK, ATM, ATR, and SMG1. ATM is the first to be recruited to DSBs, where it is activated by MRN, probably as a result of changes in chromatin configuration; it then phosphorylates and activates several DNA repair and cell cycle checkpoint proteins, i.e., the histone λH2AX and NBN, required for recruiting other ATM targets.
ATM initiates a pathway that activates NF-κB (associated with cellular survival), as well as phosphorylates BID, which plays an anti-apoptotic role.
The DNA-PK (DNA-dependent protein kinase) complex consists of a catalytic subunit (DNA-Pkcs) and a DNA-binding complex called Ku, which binds altered DNA structures such as double-strand breaks, nicks, or hairpin loops. Recombination of broken DNA strands occurs either between strands that have long stretches of homology (HR) or between DNA strand breaks, without relying on the presence of considerable homology between them. This recombination is known as nonhomologous end joining (NHEJ).138 Mutations in this system result in the accumulation of V(D)J-specific double-strand breaks, indicating a defective repair mechanism, which causes SCID. A new gene has been added to those already known that regulate double-strand DNA break repairs. It was named ARTEMIS (now known as DCLRE1C),139 after the Greek goddess who was the protector of small children and animals.140 It belongs to the superfamily of metallo-β-lactamase enzymes and is now called DNA cross-link repair K (DCLRE1C).141 It is involved in the nonhomologous end joining pathway, a DNA repair process used by eukaryotic cells,142,143 as well as in the repair of broken DNA ends from the V(D)J recombinase activity.
The repair requires that the hairpin in the coding joint is opened by the DNA-PK-DCLRE1C protein complex.141,144 In such a complex, DCLRE1C acquires endonuclease activity, which it does not possess by itself.144 DCLRE1C has been shown to be mutated in patients with SCID, expressing radiosensitivity.138 Patients with hypomorphic mutations in DCLRE1C have not only immunodeficiency, but also a predisposition to lymphoma.144
RAG1 AND RAG2 PROTEINS
The evolutionary origin of the RAG proteins has been controversial. Up to now it was accepted that the RAG genes originated from a horizontal gene transfer of a transposon, i.e., a mobile DNA element, and therefore are related to transposases, as discussed earlier.
Recently, however, two genes in sea urchins (Echinodermata) were detected that bear structural similarities to the RAG proteins.145 Echinodermata are an earlier evolutionary stage of chordates, to which vertebrates and humans belong. It is therefore possible that RAG proteins may have arisen very early in evolution and acquired their present-day function early in vertebrate evolution.
Both RAG proteins are required for V(D)J recombination, because lack of function of either one leads to SCID.146,147 A region that retains the recombinase activity is known as the core and has been used for studies in vitro. For RAG1, the core is located in the sequence 384-1,008 from a total of 1,040 amino acids, and for RAG2 in amino acids 1 to 383 of a total of 527. The core of RAG2 contains six repeats, each consisting of an antiparallel β-sheet, formed by four β-strands. The repeats are arranged in a circular formation like blades of a propeller,147 a structure that is known to mediate protein-protein interactions. The C-terminal quarter of RAG2 consists of a plant homeodomain fold, which is found in proteins with chromatin-binding properties.148
Mutational analysis of the RAG proteins has provided some clues about structure-function relations. The catalytic properties of the proteins have similarities with members of the retroviral integrase superfamily132 and require divalent metal ions for their function—a requirement that is characteristic of some nucleases, the functions of which depend on acidic amino acids. Indeed, in RAG1, several acidic amino acids are critical for both nicking and hairpin formation, without affecting the DNA binding.149 For RAG1, the N terminus is important for activity,149 and the binding of RAG1 directly to DNA is supported by basic residues of RAG2.151 Deletion of the C-terminal region of RAG2 results in a reduction of the number of B and T cells.152 The plant homeodomain of RAG2 regulates differential access to sites of recombination, and although it is dispensable for D/JH recombination, it is essential for the VH/DJH step.153
SOMATIC HYPERMUTATION
The primary importance of somatic hypermutation (SHM) is related to affinity maturation, i.e., the increase of the affinity of antibodies following repeated antigenic stimulations.154 However, a wider scope of hypermutation is to provide a better defense
against hypermutating microorganisms. Many pathogens such as Trypanosoma, Neisseria, influenza virus, and HIV evade the immune system by variations of the antigens of their coat (antigenic variation). Accordingly, SHM is the adaptation of the B cell in response to hypermutating microorganisms.155 SHM introduces mutations in the V gene at the rate of about 103 mutations/base pair/cell division, i.e., 106-fold higher than spontaneous mutations of somatic cells. At this rate genomic integrity can be maintained only if the SHM specifically targets the Ig genes. However, it has been suggested that SHM is not a mechanism specific for the Ig genes but it may occur in other genes. Perhaps the apparent preference for the Ig genes may be due to their higher density of certain hot spots,156 and SHM appears to target preferentially certain hot spots, such as the short DNA motif DGYW.
against hypermutating microorganisms. Many pathogens such as Trypanosoma, Neisseria, influenza virus, and HIV evade the immune system by variations of the antigens of their coat (antigenic variation). Accordingly, SHM is the adaptation of the B cell in response to hypermutating microorganisms.155 SHM introduces mutations in the V gene at the rate of about 103 mutations/base pair/cell division, i.e., 106-fold higher than spontaneous mutations of somatic cells. At this rate genomic integrity can be maintained only if the SHM specifically targets the Ig genes. However, it has been suggested that SHM is not a mechanism specific for the Ig genes but it may occur in other genes. Perhaps the apparent preference for the Ig genes may be due to their higher density of certain hot spots,156 and SHM appears to target preferentially certain hot spots, such as the short DNA motif DGYW.
HUMAN HEAVY- AND LIGHT-CHAIN GENE LOCI
The heavy and light chains of the Ig molecule are assembled from a large collection of preexisting gene segments by the lymphoid-specific RAG1 and RAG2 proteins. The RAG proteins cooperate to make double-strand breaks at specific recognition sequences (recombination signal sequences) or RSS.157 The recombination takes place between one RSS with 12-bp spacer (12-signal) and one with a 23-bp spacer (23-signal) and this arrangement is called the 12/23 rule. In the presence of the metal ion Mn2+, a single signal sequence supports double-strand cleavage, whereas Mg-2+ requires two signal sequences.157 The two light-chain isotypes, κ (kappa) and λ (lambda), comprise approximately 60% and 40% of all igs, respectively, and consist of a V domain and a C domain of approximately 107 residues each in length. The V domain is encoded by the V and J segments, the former encoding the first 95 to 96 residues and the latter encoding the remaining 12 to 13. There is a single C-κ gene, and several C-λ genes. The κ locus contains approximately seventy-six V-κ segments grouped into six families, and five J-κ segments, but no d segments. The λ-locus contains 10 families of V-λ segments, a high number of pseudogenes, and several C-λ genes, each preceded by a single J-λ segment.
One of the consequences of the imprecision of the V(D)G recombination is a change in the reading frame at the junction between the two gene segments, causing the segments to join out of phase, so that the triplet reading frame for translation is not preserved. In this case the rearrangement results in V(D) J combinations with numerous stop codons which interrupt the translation. Recombination may be nonproductive or productive; when the junction lies within a codon, the resulting amino acid is encoded by nucleotides from both gene segments involved. It is estimated that only one in three recombination attempts are productive, but the imprecision in the joints between variable gene segments increases their diversity by at least 100-fold. Difference in mRNA stability allows pro-B cells to distinguish between productive and nonproductive Ig gene rearrangements.
The Ig gene rearrangements follow a certain sequence of the various loci and use only some of the available gene segments (allelic exclusion), which indicates that there are regulatory mechanisms underlying the process of rearrangement. For the IgG molecule the IGH@ gene is rearranged first and forms a complete V gene from three individual segment clusters, i.e., V, D, and J. One of the D segments joins a JH segment forming the D-JH complex and in the next step, which is regulated by the Dµ protein, the V segment joins the complex. A D-JH rearrangement has also been found sometimes in T cells. The Dµ protein, which lacks a VH segment, is encoded by the D-JH complex and the constant gene of the µ-heavy chain and may be expressed on the membrane of the cell with an SLC. Expression of Dµ protein prevents further VH-to-DJH rearrangement, or the rearrangements are diverted to another pathway such as κ-chain rearrangements, which are accelerated. Completion of the µ-chain inhibits further rearrangements by the opposite allele (known as allelic exclusion).158 Completion of the µ-chain rearrangement is followed by rearrangements of the IGK@ gene, and if both κ alleles fail to rearrange productively, the IGL@ gene is rearranged (Fig. 12.6). However, this hierarchical order of Ig gene rearrangements has been challenged, because examination of B cells at different stages of early development shows that the heavy and light-chain genes rearrange independently.159 At the end of its early developmental stage, the B cell emerges with the expression on its surface of a unique antigen receptor consisting of IgM accompanied by two accessory molecules, CD79A and CD79B.160
REGULATION OF V(D)J RECOMBINATION
Initiation of rearrangements by the recombinase requires that the gene must be accessible, i.e., the locus must be able to act as a template for the recombinase. In cells competent for rearrangement it was found that JK and VH genes have already been transcribed, but these RNAs, named germline or sterile transcripts, are incapable of encoding the protein. Several possibilities may exist to explain this finding, and since inactive genes are not accessible to the recombinase, it is possible that the altered structure of the chromatin allows the recombinase to recognize the RSS. Alternately, the enhancers may establish altered chromatin regions where both the recombinase and the transcription machinery have access; this is known as the accessibility hypothesis.104,161 Enhancers and promoters function as accessibility control elements (ACEs), which regulate V(D)J recombination. Enhancers maintain an open chromatin state in multiple embryonic tissues independent of their activity level. Regions of nonprotein coding genomic DNA bind protein transcription factors (TFs) and thus direct the expression of target genes. The enhancer of the IgM heavy chain (Eµ) is located within the intron between the JH segments and the Cµ gene and is associated with matrix attachment regions (MAR), which improve production of the protein by recombination.162 Transcriptional regulation by enhancers is fundamental to the evolutionary diversity of metazoans as well as their embryonic development. The regulation of a gene is an exceedingly complex process controlled by interacting proximal and distal DNA elements usually placed in a cis configuration, proximal to the basal promoter where the general transcription machinery assembles. A promoter is always located in close proximity to the 5′-end of a gene and is necessary, but not sufficient, for its transcription, while enhancers or silencers are distant elements.
Enhancers are thought to be composed of binding sites for transcription factors (TF), which upon recruitment to the enhancer loop over the promoter and activate the transcription of the target gene. There are three additional enhancers within the Ig heavy-chain locus, and one of them (DQ52) competes more efficiently in conferring accessibility to the JH region. Interaction between DQ52 and Eµ is likely responsible for ordered rearrangement, i.e., D to JH followed by VH-to-DJH complex.163 The κ-light chain intronic enhancer is active only in mature B cells and plasmacytomas, and its stage restriction is dictated by a single motif that binds the transcription of nuclear factor-κB (NF-κB), which recognizes a 10-bp motif. The NF-κB consists of two subunits and the DNA-binding function is mediated by the smaller one. The enhancers for the Ig transcription lie in the vicinity of the C-α gene and κ locus. The enhancer between the Jκ and Cκ genes becomes transcriptionally active in the transition from the pre-B to B-cell stage.164 The matrix attachment regions are regions rich in AT
and demarcate regions of chromatin that undergo base unpairing and other regions that mediate binding of topoisomerase II. This regional division of chromatin may also be regulated from a distance by trans-acting factors, which bind to specific motifs within the enhancer or promoter. Despite the promiscuity and redundancy of regulatory sequences, transcription is still specific for tissue lineage and even stage of development. Several transcription factors and other signals lead to changes in expression levels, yet no clear model exists to explain the complexity of transcriptional regulation. The clear result is that disruption of the TCF3 and EBF1 transcription factors results in blocking of B-cell development at a stage before Ig heavy-chain gene rearrangements.
and demarcate regions of chromatin that undergo base unpairing and other regions that mediate binding of topoisomerase II. This regional division of chromatin may also be regulated from a distance by trans-acting factors, which bind to specific motifs within the enhancer or promoter. Despite the promiscuity and redundancy of regulatory sequences, transcription is still specific for tissue lineage and even stage of development. Several transcription factors and other signals lead to changes in expression levels, yet no clear model exists to explain the complexity of transcriptional regulation. The clear result is that disruption of the TCF3 and EBF1 transcription factors results in blocking of B-cell development at a stage before Ig heavy-chain gene rearrangements.
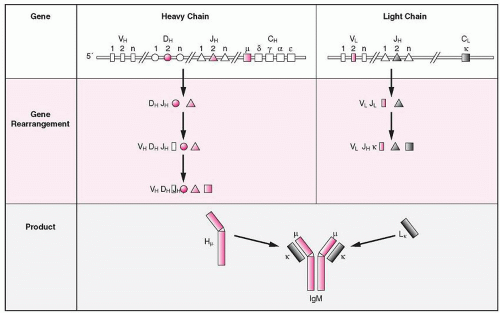 FIGURE 12.6. Heavy-chain immunoglobulin (Ig) M and kappa light-chain rearrangement. With the completion of the V(D)J recombination of the µ chain and the formation of a complete µH chain, the LK chain rearranges, and, if successful, it forms the complete IgMK molecule. If both κ alleles rearrange unsuccessfully, the λ gene rearranges. If successful, an IgMλ molecule is formed. If both λ genes cannot rearrange successfully, the cell dies by apoptosis. |
BCR V GENE REPERTOIRE
Certain aspects of the study of the Ig V gene repertoire and the Ig gene rearrangement have found wide applications in clinical medicine.165 The total number of V genes defines the available repertoire. However, not all genes are equally functional, and as a result, the repertoire which is usually expressed is biased, because some of the V genes are expressed with a significantly higher frequency than would be expected if all had an equal chance for rearrangement. This biased expression affects all V, D, and J gene segments; thus the JH4 segment (one of the existing six), on the basis of equal opportunity among all five segments, should be detected in 17% of B cells, yet it is found in 32% of B clones from fetal liver and in 42% of pre-B-cell ALL. The bias of the expressed repertoire shows striking association with certain diseases. For example, the VH4-21, a member of the VH4 family, is found in cold agglutinins.166 Preferential expression of certain VH segments is also detected in ALL clones. In ALL the frequency of N-nucleotide additions differs in children less than 3 years of age (12.5%) compared with children greater than 3 years old (89%).167 Because in fetal B lymphocytes the frequency of N-nucleotide additions is low, it is suggested that the transforming event in the younger age group probably occurred during fetal life.
In chronic lymphocytic leukemia (CLL), the use of V genes is also restricted, and the same is true in non-Hodgkin lymphomas. It is interesting that in certain cases of follicular lymphoma, the V genes not only have undergone somatic mutations, but also continue to do so during the course of the disease, suggesting that the malignant clone is responsive to an antigen.168,169 Somatic mutations, as expected, have also been detected in multiple myeloma, because the malignancy derives from an advanced stage of differentiation of B cells.170 This brief summary emphasizes the fact that defects in V(D)J recombination171 are associated with human diseases such as loss of RAG1/RAG2 function, which results in SCID. In certain autosomal recessive diseases, hypersensitivity to DNA-damaging agents results in chromosomal breaks and rearrangements, with an increased incidence for development of leukemia and other malignancies. They have been classified as DNA repair disorders and include AT, Fanconi anemia, xeroderma pigmentosum, and Bloom syndrome (see later). Genomic instability is most often linked to DNA repair deficiencies, which suggests that evaluation of deficiencies in repair mechanisms in pluripotent human stem cells should be a necessary step prior to their clinical and research use. The DNA repair pathways and repair capacities in pluripotent cells are more heterogeneous than those of differentiated cell lines. In mice the SCID mutation, an autosomal recessive mutation, results in the absence of B and T cells as a result of impairment of the V(D)J recombination. Both lymphoid and nonlymphoid SCID cells are hypersensitive to killing by ionizing radiation because of a DNA double-strand base-repair
defect. It is rather important that a human gene on chromosome 8q11 has been identified that restores V(D)J recombination, double-strand base repairs, and normal resistance to irradiation in SCID cells.172,173 V(D)J recombination is necessary for immune diversity and survival, but this objective can be achieved only through DSBs which threaten genomic stability. The complexity of the recombinase function is matched by complex DNA repair machinery. Disorders related to V(D)J recombination may be considered in two categories: those related to the first step (cleavage) and those related to the second step (cleavage repair).174
defect. It is rather important that a human gene on chromosome 8q11 has been identified that restores V(D)J recombination, double-strand base repairs, and normal resistance to irradiation in SCID cells.172,173 V(D)J recombination is necessary for immune diversity and survival, but this objective can be achieved only through DSBs which threaten genomic stability. The complexity of the recombinase function is matched by complex DNA repair machinery. Disorders related to V(D)J recombination may be considered in two categories: those related to the first step (cleavage) and those related to the second step (cleavage repair).174
THE MUTATION IN SEVERE COMBINED IMMUNODEFICIENCY
Two fundamental processes operate in V(D)J recombination, i.e., double-strand breaks during the gene rearrangements, followed by DNA repair. The SCID mutation in mice175 is associated with lack of mature lymphocytes and agammaglobulinemia. SCID T cells have long P-nucleotide sequences and accumulate hairpins at the TRD@ coding ends. In addition to the lack of rearrangements, there is an inability to repair damage from irradiation. In mice the SCID mutation is located on chromosome 16, and since human chromosome 8 can complement mouse SCID defects, it has been assumed that the SCID defect for humans should be in chromosome 8, although human chromosome 8 is not associated with any known immunodeficiency or DNA repair diseases. The gene, however, for the DNA polymerase-8, is in chromosome 8p11-12. Mutated RAG genes have been detected in nine patients from seven families.176 Recombination activity was undetectable with mutations in the RAG core domains, and in four patients there was a complete B-cell differentiation arrest between the pre-BI and pre-BII stages.
V(D)J RECOMBINATION: DNA CLEAVAGE
Two types of RAG1 and RAG2 gene inactivations have been identified in mice. The first type results in the inactivation of the recombinase so that V(D)J recombinations cannot be initiated,108,109 and as result the mice have no mature T and B lymphocytes, although there is an increase of immature lymphocytes in the lymphoid organs. The second type of mutation consists of a single conservative amino acid substitution in both proteins. It does not affect the initiation of V(D)J recombination, but severely impairs the coding and signal joint formation.177,178 The mutant enzymes are defective in hairpin opening in vitro and the formation of the coding joint in vivo. This evidence indicates that the RAG proteins, in addition to their cleavage function, are important for the formation of the joints; therefore, they must remain associated with the postcleavage complex to achieve opening of the hairpin.
In severe combined immune deficiency (SCID), approximately 20% of the patients lack both T and B lymphocytes, but have normal NK cell counts;179 some of these patients have RAG1 or RAG2 gene mutations,110 while others have increased radiosensitivity of their bone marrow cells and fibroblasts (RS-SCID).180 The RAG2 structure consists of two globular domains separated by a hinge of approximately 60 amino acids. The largest domain which is enzymatically active has 350 residues and includes the previously described core region of RAG2. This domain forms a β-propeller with six blades arranged in a circle from a center. The β-propellers are made of 4 to 10 β-sheet units (blades) circularly arranged around a central tunnel, and have probably evolved by duplication and fusion of a rudimentary repetitive unit. The beta-propeller fold is phylogenetically widespread and able to support a variety of functions such as catalysis, ligand binding and transport, regulation, and protein binding. Interestingly, it appears that the beta-propeller topology is also compatible with strikingly diverse sequences. Amongst this diversity there are three large groups of proteins with related sequences and very important cellular and intercellular regulatory functions. The WD-40 repeat proteins have a variety of functions, such as signal transduction, transcription regulation, cell cycle control, autophagy, and apoptosis. The YWTD proteins and the Kelch repeats interact directly with the actin-binding protein LASP1 in membrane ruffles at the tips of pseudopodia, where both proteins are necessary for pseudopodial elongation. A common characteristic among the proteins of these families is the internal repeats of 40 to 45 residues. The β-propellers are widely utilized in nature as recognition modules, and the well-conserved β-propeller fold exhibits a high degree of functional diversity, which is probably accomplished through variations in the surface properties of the proteins.181 The β-propeller fold shows a high degree of functional diversity, probably accomplished through variations in the surface properties of the proteins. It is present in some integrins and other proteins which are known to mediate protein-protein interactions. Six out of seven mutations so far described in RAG2 in humans are clustered on one side of the propeller. This side is involved in interactions between RAG1 and RAG2 which are critical for functional activity. Two of these mutants (C41W and M285R) reduce the interaction between RAG1 and RAG2 in vitro but no DNA-binding activity of the complex was detected.182 This is consistent with the hypothesis that RAG2 stabilizes the RAG1/RAG2 complex.183
OMENN SYNDROME
Omenn syndrome (OS) is an inherited disorder characterized by absence of circulating B cells, infiltration of the skin and intestine with activated oligoclonal T lymphocytes (HLA-D+), eosinophilia, and elevated serum IgE.184
The immunologic hallmark of OS is the expansion and activation of an oligoclonal population of autoreactive T cells. These cells should be controlled rapidly by immunosuppressive agents.185 The patient has diffuse erythroderma, lymphadenopathy, hepatosplenomegaly, protracted diarrhea, and failure to thrive. The T cells are activated, secrete Th-2 types of cytokines,186 and express oligoclonal expansion of TRB@ chains with VDJ junction similarities, suggesting a common antigenic specificity with possible autoimmune proliferation and a critical defect in the development of lymphocytes.187 The hallmarks of OS are expansion and activation of an oligoclonal population of autoreactive T cells, skin erythroderma, and elevated IgE in the absence of circulating B cells.188 The syndrome expresses clinical symptoms and signs which define an inflammatory condition associated with various genetic defects.188 The patients have activated oligoclonal T cells, and it appears that genes other than those directly involved in V(D)J recombination may play a role in the pathogenesis of OS. Omenn-like features are associated with mutations of genes involved in the maturation steps of lymphoid cells and not those directly regulating V(D)J recombination.188
OS is characterized by a cellular infiltrate of the skin, gut, liver, and spleen and a resemblance to graft-versus-host-like disease. The V(D)J recombination process is impaired but not abolished and leads to the generation of a few T cells which expand in the periphery and infiltrate target organs such as skin and gut, resulting in severe erythroderma and colitis, supporting the evidence that Omenn is an inflammatory condition.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


