FIGURE 102-1. Age distribution in adrenal Cushing’s syndrome according to etiology, depicted as percentage of patients at different ages: adenoma (-◊-), carcinoma (-•-), PPNAD (…….), and AIMAH (- – – ).
Adrenal adenoma occurs mostly in females, as does—to a lesser extent—cortisol-secreting adrenal cancer.5,18,19 No clear-cut sex-related pattern has been observed for Cushing’s associated with AIMAH and PPNAD.
Mortality from untreated Cushing’s syndrome is high, with 50% survival 5 years after the diagnosis,20 mostly due to cerebrovascular events and infections. Upon removal of a benign adenoma, taking perisurgical mortality into consideration, 5-year survival averaged 77%21 in the 1980s, but is nowadays comparable to mortality in the general population.5,22 As regards mortality from malignant adrenocortical cancer, outcome is dismal, with high mortality for stage III and IV disease and slightly better survival for patients diagnosed with localized disease.23–25 In Cushing’s syndrome due to PPNAD within Carney’s complex, a familial multiple endocrine neoplasia syndrome, extraendocrine manifestations such as cardiac myxomas or malignant Schwannomas contribute to excess mortality.26
Clinical Features of Hypercortisolism
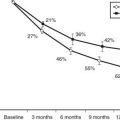
Cushing’s syndrome features all the consequences of tissue exposure to excessive cortisol concentrations, including aspecific alterations such as obesity, hypertension, and mood changes, and more unique signs such as proximal muscle wasting, purple striae, and easy bruising (see Chapter 15). The severity of clinical manifestations varies according to the extent and duration of cortisol hypersecretion, with adrenal cancer characterized by rapid attainment of very high cortisol levels and severe clinical signs; adenomas present milder glucocorticoid hypersecretion and a more indolent course. In fact, the clinical presentation of adrenal adenomas closely resembles that of pituitary-driven Cushing’s disease, whereas adrenal cancer mimics ectopic ACTH secretion due to poorly differentiated neuroendocrine tumors. On average, over 2 years are necessary for a cortisol-secreting adrenal adenoma to be diagnosed after the appearance of the first symptoms, but the remarkable clinical presentation of adrenal carcinoma leads to the diagnosis in half that time.7,27 Indeed, the time to diagnosis is inversely related to mean cortisol levels (Fig. 102-2).
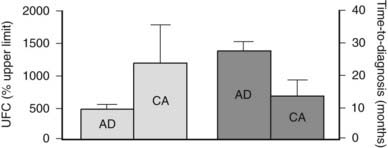
FIGURE 102-2. Urinary free cortisol (UFC) and time to diagnosis in patients with Cushing’s syndrome due to adrenal adenoma (AD) or adrenal carcinoma (CA). UFC is expressed as percent of the upper limit of the normal range.
Specifically, patients with hypercortisolism usually present with central obesity, a round and erythematous face, supraclavicular fat pads, cervical fat accumulation (the so-called “buffalo hump”), thinning of the skin, purple striae, and proximal muscle weakness. Hirsutism, a common feature of ACTH-dependent Cushing’s syndrome, is usually absent in benign tumors that secrete only cortisol but, conversely, accompanies malignant transformation.7 Hypertension as well as osteopenia and pathologic fractures (ribs, feet, and vertebrae are frequently affected sites) are common, and osteopenia in young patients with Cushing’s syndrome should raise the suspicion for PPNAD (see later). Derangements in blood chemistry include increased white blood cell count with granulocytosis, lymphopenia, and eosinopenia; a slightly elevated red blood cell count; increased VLDL, LDL, and HDL cholesterol and triglycerides; and frequently, impaired glucose tolerance or overt type 2 diabetes mellitus. Electrolyte imbalances such as hypokalemia are more common in severe hypercortisolism. Case history will reveal menstrual irregularities or even amenorrhea, reduced libido or impotence, fatigue, easy bruising, insomnia, and psychiatric alterations. These latter range from changes in mood, irritability with rage fits, anxiety, impaired memory, and concentration to psychosis with frank depression and manic episodes. Curiously, in children and adolescents, hypercortisolism has been associated with overachievement both in school and in extracurricular activities. Less frequent signs are cutaneous fungal infections, wound dehiscence, exophthalmos, nephrolithiasis, glaucoma, and headache. As mentioned before, severity of the clinical presentation often reflects the underlying etiology and features such as extreme muscle atrophy, hypokalemia, hypercalcemia, and the abrupt appearance of diabetes should increase the suspicion of adrenocortical cancer (Fig. 102-3). Except for signs of virilization, however, no single sign or symptom is significantly overrepresented in either etiology.7,16 The hormonal secretory pattern of adrenal carcinoma may change over time. Curiously, Cushing’s syndrome developed in two previously asymptomatic patients after recurrence of adrenocortical carcinoma, but conversely, no sign of hypercortisolism accompanied relapse of a previously symptomatic adrenocortical carcinoma.18
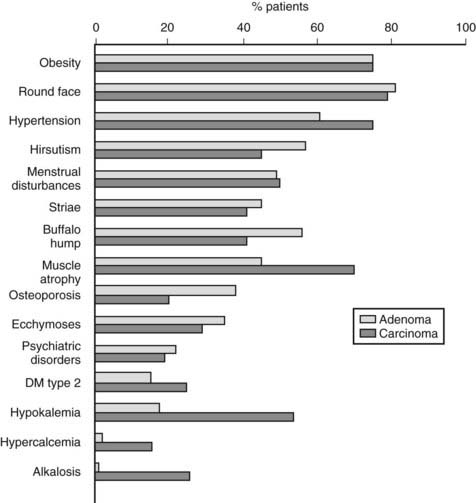
FIGURE 102-3. Clinical presentation in patients with Cushing’s syndrome due to adrenal adenoma or adrenal carcinoma.
In addition to cortisol hypersecretion, adrenal androgens may also be produced in excess, most notably in adrenocortical cancer and more rarely in adenomas. Androgen secretion reflects inefficient steroidogenesis, since enzymes involved in the later steps of cortisol synthesis (e.g., 11β-hydroxylase or 21β-hydroxylase) are unable to transform their substrate overload. Precursors, therefore, accumulate and are shunted to alternative biosynthetic pathways, leading to accumulation of DHEA, DHEAS, androstenedione, and estrogen (see Chapter 96). This alteration is the biochemical correlate of progressive dedifferentiation of adrenal tumors, with steroidogenesis proceeding to its final product, cortisol, in benign and hyperplastic lesions, whereas poorly differentiated carcinomas are unable to efficiently carry steroidogenesis to term.28 From a clinical viewpoint, androgen hypersecretion gives rise to significant hirsutism, acne, and temporal balding in adult patients and contributes to menstrual alterations in women. In children, hyperandrogenism may arrest linear growth in boys or lead to sexual precocity and virilization in girls.29 Excess estrogen secretion may lead to gynecomastia in men.30 Mixed cortisol- and aldosterone-producing adenomas have also been reported,31 as have mixed cortisol-, androgen-, and estrogen-producing adenomas.32
Adrenal cortex carcinomas may present with mass-related signs, symptoms of metastatic disease, or nonspecific features of malignancy such as weight loss, fever, and anorexia. Palpable and painful abdominal masses, an acute abdomen, and vascular obstruction by tumor thrombi of hepatic veins (Budd-Chiari syndrome) or the inferior vena cava may first raise suspicion of adrenal cancer. Metastases occur most frequently to liver, lungs, retroperitoneal lymph nodes, and bone and may give rise to back pain and osteoporotic fractures, or hypoglycemia if the liver is extensively infiltrated18,24 (see Chapter 106).
The clinical picture of the other etiologies of adrenal Cushing’s syndrome, AIMAH and PPNAD, presents certain unique features such as overt hypercortisolism only during particular phases of life—for example, pregnancy and menopause in luteinizing hormone (LH)-dependent AIMAH or signs of Carney’s complex in familial PPNAD. These issues will be discussed in more detail in specific sections (see later).
Diagnosis
DIAGNOSIS OF CUSHING’S SYNDROME
The diagnosis of Cushing’s syndrome due to adrenal tumors is first raised by the clinical presentation then confirmed by both biochemical and radiologic investigations. Hormonal measurements reflect the functional derangement of the hypothalamo-pituitary-adrenal (HPA) axis consequent to sustained autonomous cortisol production by the adrenal gland (see Chapter 15). Two features of the HPA axis are relevant to pathophysiology of primary adrenal hypercortisolism: (1) homeostasis of the HPA axis is assured through long and short feedback loops acting at hypothalamic (i.e., corticotropin-releasing hormone [CRH]), pituitary (i.e., ACTH), and adrenal (i.e., cortisol) levels, with cortisol levels as the effector of feedback; (2) cortisol and ACTH are secreted in pulsatile fashion, inscribed within a circadian rhythm with high secretory output in the early morning, progressive decrease during the day, and nadir around midnight. If adrenal cortisol production is autonomous, as occurs in adrenal-derived Cushing’s syndrome, high steroid secretion will activate negative-feedback loops leading to suppression of hypothalamic and pituitary secretion and thus disruption of physiologic pulsatility and circadian rhythmicity of ACTH and cortisol secretion. The HPA axis will also be resistant to further inhibition by exogenous steroids. The diagnosis of Cushing’s syndrome is performed by looking for these alterations: evaluation of integrated daily cortisol secretion and its circadian rhythmicity and suppressibility with dexamethasone. Details of these investigations are given elsewhere (see Chapter 15), but specific changes expected in adrenal-driven disease are given in following discussions.
Urinary free cortisol (UFC) levels are usually very high in patients with adrenocortical cancer (up to 100 times greater than normal), whereas such striking elevations are less frequent in patients with cortisol-secreting adenoma (Fig. 102-4). Indeed, UFC concentrations may occasionally fall within the normal range in these latter patients,27 and this underlines the need for multiple UFC measurements.33,34 In patients with AIMAH, UFC concentrations are rarely very high and may fluctuate according to stimulation by the causative aberrant receptor. For example, patients in whom hypercortisolism was dependent upon adrenal cortex stimulation by LH, who exhibit markedly elevated UFC concentrations and develop Cushingoid features only during pregnancy and menopause.35,36 In young patients with PPNAD, again, UFC rarely reaches very high levels and may present slow, periodic progression.12,37,38 Other tests usually involve assessment of cortisol daily variations or its suppressibility by dexamethasone. In patients with full blown hypercortisolism, two clearly pathologic results or even one are sufficient to establish the diagnosis of Cushing’s syndrome and proceed with the diagnostic workup. However, in patients with mildly elevated or fluctuating cortisol values (as often occurs with AIMAH or even PPNAD), repeat evaluations over time are necessary. A secure diagnosis of Cushing’s syndrome is essential before performing tests for the etiologic diagnosis of hypercortisolism, because the results obtained with these procedures may overlap with normal physiology.
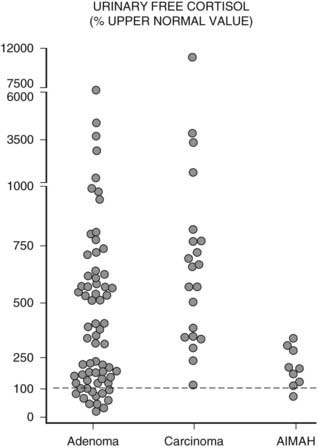
FIGURE 102-4. Distribution of urinary free cortisol (UFC) levels in patients with adrenal Cushing’s syndrome. Values are expressed as percentage of the upper limit of normal range (dashed line = 100%).
(Adapted from Invitti C, Pecori Giraldi F, De Martin M, Cavagnini F: Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study, J Clin Endocrinol Metab 84:442, 1999. © Endocrine Society.)
DIAGNOSIS OF ADRENAL CUSHING’S SYNDROME
Once the existence of Cushing’s syndrome is firmly established, the etiology of the disease has to be defined. In adrenal Cushing’s syndrome of any given etiology, autonomous cortisol secretion by the adrenals feeds back at hypothalamo-pituitary level to inhibit CRH and ACTH secretion. Plasma ACTH levels will therefore be suppressed in adrenal Cushing’s syndrome, whereas inappropriately high ACTH concentrations characterize ACTH-dependent Cushing’s syndrome (i.e., pituitary or ectopic ACTH secreting tumor; see Chapter 15). Measurement of plasma ACTH levels is therefore pivotal to the differentiation of ACTH-dependent from ACTH-independent Cushing’s syndrome. As mentioned above, ACTH is secreted in pulsatile fashion, dictating the same circadian rhythm to cortisol. Further, ACTH is extremely sensitive to stress, has a short plasma half life, and is easily degraded by plasma peptidases. For these reasons, blood samples for ACTH measurement have to be collected in unstressed conditions, preferably two or three samples over 30 minutes, into tubes containing enzyme inhibitors and conserved at low temperatures prior to separation into aliquots. Repeated freeze/thaw cycles should be avoided.
In the vast majority of patients with adrenal Cushing’s syndrome, plasma ACTH concentrations are suppressed, thus providing clear distinction from ACTH-dependent Cushing’s syndrome. Initially formulated guidelines indicated that morning ACTH concentrations lower than 10 pg/mL (2 pmol/L) or evening ACTH levels lower than 5 pg/mL (1 pmol/L) were indicative of ACTH-independent Cushing’s syndrome.39 In RIA assays with 5 to 10 pg/mL sensitivity, this corresponds to values below the detection limit. With the newly developed IRMA or chemiluminescent assays capable of measuring plasma ACTH concentrations as low as 1 pg/mL (0.2 pmol/L), ACTH values may be detectable in patients with adrenal Cushing’s syndrome but usually are below the lower limit of the normal range.16,27,40,41 However, in an Italian study on over 50 patients with adrenal Cushing’s syndrome, up to one fourth presented ACTH concentrations within the normal range27 (Fig. 102-5), but whether this is because of issues in assay methodology, as seems more likely, or to incomplete suppression of pituitary ACTH secretion remains to be established. Occasionally, ACTH concentrations are frankly non-suppressed40,42,43; in one patient bearing an adrenal carcinoma, ACTH concentrations were persistently high because the tumor itself produced ACTH.44 In addition to patients with adrenal Cushing’s syndrome presenting measurable or normal ACTH concentrations, some 10% of patients with Cushing’s disease display ACTH values below 10 pg/mL (2 pmol/L),27 thus blurring the boundaries between the two etiologies. To correctly diagnose patients falling into this grey area, stimulation with CRH proves of use; patients with pituitary-dependent hypercortisolism will mount an ACTH response to the stimulus, whereas patients with ACTH-independent hypercortisolism will not27,45,46 (Fig. 102-6).
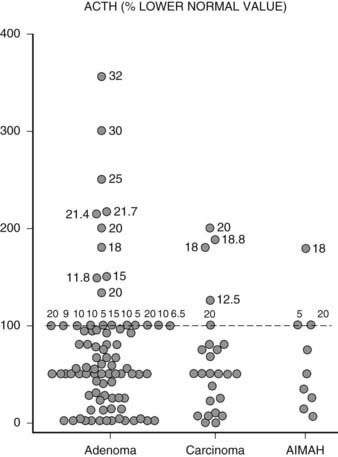
FIGURE 102-5. Distribution of plasma ACTH levels in patients with adrenal Cushing’s syndrome. Values are expressed as percentage of the lower limit of the normal range. For values within the normal range, numbers close to individual points indicate absolute ACTH concentrations in pg/mL measured using different assays.
(Adapted from Invitti C, Pecori Giraldi F, De Martin M, Cavagnini F: Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study, J Clin Endocrinol Metab 84:442, 1999. © Endocrine Society.)
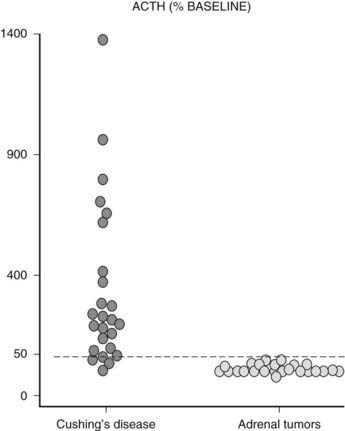
FIGURE 102-6. ACTH response (% baseline) to stimulation with CRH in patients with Cushing’s syndrome presenting with adrenal nodules.
In the past, tests such as stimulation with metyrapone or suppression with high-dose dexamethasone have been employed to distinguish between adrenal and pituitary Cushing’s syndrome,47 but their suboptimal specificity has superseded their routine use for this purpose. However, the high-dose dexamethasone suppression test has been re-proposed for the etiologic diagnosis of adrenal Cushing’s syndrome (see later).
DIFFERENTIAL DIAGNOSIS OF ADRENAL CAUSES OF CUSHING’S SYNDROME
The etiology of adrenal Cushing’s syndrome is usually established by imaging of the adrenals (see also Chapter 105), since biochemical and clinical features overlap considerably between adenoma, carcinoma, and adrenal hyperplasia.7,14,16 Hormonal measurements provide useful adjunctive information but rarely establish the diagnosis with certainty. Genetic causes, such as PPNAD or MAS, may be recognized by features typical to the syndrome (e.g., lentigines or myxomas and fibrous osseous dysplasia, respectively; see later).
Steroid Markers
Dehydroepiandrosterone Sulfate
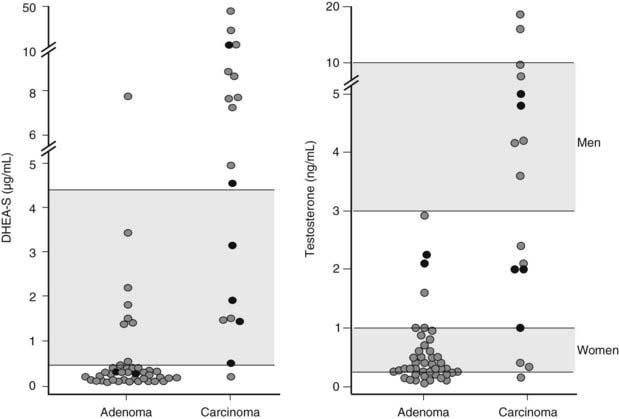
Measurement of serum dehydroepiandrosterone sulfate (DHEAS) may aid in the distinction between benign and malignant adrenal tumors. Indeed, although no cutoff value ensures absolute diagnostic accuracy, low age-and-gender-adjusted DHEAS levels are usually found in Cushing’s syndrome due to adrenal adenoma, whereas DHEAS levels are markedly increased in adrenal carcinoma (Fig. 102-7), especially in children.16,29,48,49 Two mechanisms underlie this diversity: on the one hand, DHEAS is an ACTH-dependent androgen, so inhibition of the normal adrenal gland in patients with cortisol-secreting adrenal adenoma will lead to reduced DHEAS secretion. On the other hand, inefficient steroidogenesis in malignant neoplasms leads to the release of several steroid precursors, including delta-5 steroids such as DHEAS. Of interest, DHEAS levels often remain suppressed long after removal of the hypersecreting adrenal gland, occasionally up to 8 years after surgery.48,50 This is in contrast with the recovery of ACTH and cortisol secretion, which usually occurs 12 to 18 months after surgery and indicates a dissociation of adrenal DHEAS and cortisol regulation.48,50 Enhanced sensitivity of the reticular zone to ACTH deficiency and preferential inhibition of 17,20-lyase in the atrophic adrenal gland have been hypothesized as causal factors.
Testosterone and Androstenedione
Plasma testosterone levels are within the lower centiles of the normal range in men with cortisol-secreting adrenal adenoma,51 reflecting testosterone suppression (see Fig. 102-7). This is in line with the 30% to 50% fall in testosterone levels reported after administration of high doses of cortisol or dexamethasone52 and its rebound increase following removal of the adenoma.51 Normal testosterone levels have been reported in women with cortisol-secreting adrenal adenomas7 (see Fig. 102-7). Androstenedione levels are mostly low-normal in patients of both sexes with cortisol-secreting adrenal adenomas.29,53 Conversely, in adrenal carcinoma, androstenedione and testosterone levels may be high in both men and women (see Fig. 102-7) when impairment of downstream steroidogenic enzymes in the tumor, in particular 21-hydroxylase and 11β-hydroxylase, leads to accumulation of progesterone or pregnenolone and shunting of these precursors towards 17,20-lyase53 (see Figure in Chapter 96). Accumulated DHEA can be transformed into androstenedione and subsequently into testosterone. Indeed, the detection of elevated testosterone or androstenedione in a patient with an adrenal tumor is virtually diagnostic for adrenal carcinoma.29,53 In children with adrenal carcinoma, Cushing’s syndrome is often accompanied by virilization, and testosterone, androstenedione, and DHEAS levels may reach extremely high levels.24,29
17-Hydroxyprogesterone and 11-Deoxycortisol
These two cortisol precursors are usually normal in patients with benign cortisol-secreting adrenal lesions and elevated in malignant adrenocortical tumors.24,29,53 Normal plasma hormone levels, however, do not allow adrenocortical cancer to be ruled out.
Urinary Steroid Profile
The hallmark of hypercortisolism is excess urinary excretion of cortisol and its main metabolites, such as tetrahydrocortisol, tetrahydro-11-deoxycortisol, tetrahydrocortisone and derivate cortols and cortolones (see Chapter 96). In addition, minor cortisol metabolic pathways, such as 6β-hydroxylation, are recruited to metabolize excess cortisol. In cortisol-producing adenomas, steroid synthesis is efficient, and only minor amounts of early products of the steroid cascade are excreted in urine.54,55 Conversely, in adrenocortical carcinoma, steroid precursors accumulate, and 3β-hydroxysteroids, desoxycorticosterone, and pregnanetriol—a 17-hydroxyprogesterone metabolite—become abundant in urine.54,56 Urinary 3β-hydroxy DHEA metabolites, such as 16alpha DHEA, androstenediol, and androstenetriol, as well as androstenedione-derived products such as etiocholanolone, androsterone, and androstanediols (Fig. 102-8), are usually high in adrenocortical carcinomas.55,57 In addition to these metabolites of early steroid synthesis products, tetrahydro-11-deoxycortisol, the metabolite of the cortisol substrate, is also typically increased in adrenocortical carcinoma, a consequence of relative 11β-hydroxylase deficiency.55,56 Investigation of the urinary steroid profile by gas chromatography mass spectrometry may reveal unique steroid excretory patterns, but the wide variability of excreted steroids and its changes over time hamper its routine use in clinical setting.
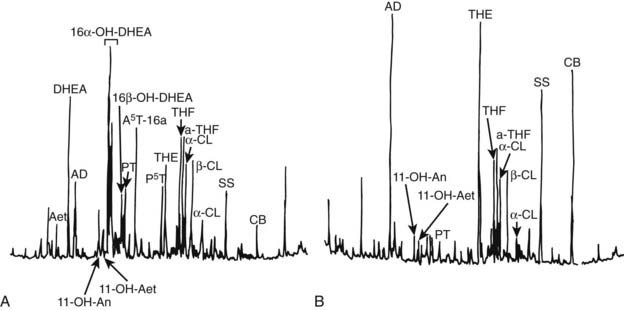
FIGURE 102-8. Chromatographic profile of urinary steroids in (A) a 6-year-old girl bearing an adrenal carcinoma and (B) an age- and sex-matched normal subject. Androsterone (An), etiocholanolone (Aet), androsten-3β, 16α, 17β-triol (A5T), pregnanetriol (PT), pregnentriol-5-ene-3βhydroxy (P5T), tetrahydrocortisone (THE), tetrahydrocortisol (THF), allo-tetrahydrocortisol (a-THF), cortols (C), cortolones (CL); internal standards (AD, SS, CB).
(From Homoki J, Holl R, Teller WM: Urinary steroid profiles in Cushing’s syndrome and tumors of the adrenal cortex, Klin Wochensch 65:721, Fig. 1, 1987; © Springer).
High-Dose Dexamethasone Testing
Adrenal tumors, both benign and malignant, fail to suppress UFC secretion after 2 days of 2 mg of oral dexamethasone every 6 hours; cutoffs for suppression are a 50% decrease from baseline of UFC levels. Patients with PPNAD often exhibit a marked paradoxical rise in UFC levels during high-dose dexamethasone testing.15,37,41 This test falls short of providing diagnostic certainty in the differential diagnosis of adrenal Cushing’s syndrome, since some patients with adrenal adenoma also exhibit an increase in UFC levels after 8 mg dexamethasone for 2 days,15,27,37 but a doubling of UFC levels is highly suggestive of PPNAD. Parallel evaluation of creatinine clearance should be performed to exclude gross changes in glomerular filtration rate.58 However, patients with malignancy may also exhibit a paradoxical increase in cortisol levels after high-dose dexamethasone testing,43,59 so this procedure should be performed only after imaging has narrowed down the choice to benign adrenal lesions.
Stimulation of Aberrant Adrenal Receptors
These procedures are useful if the history suggests hypercortisolism associated with given conditions (e.g., pregnancy, meals) or in the presence of patients with AIMAH. For a detailed discussion, the reader is referred to the heading ACTH-Independent Adrenal Hyperplasia.
Subclinical Cushing’s Syndrome
This condition, characterized by subtle alterations of HPA-axis parameters in patients who do not present stigmata of Cushing’s syndrome, is being increasingly recognized in subjects with an adrenal incidentaloma. Indeed, a variable percentage of adrenal incidentalomas will reveal themselves as cortisol-secreting adenomas, 5% to 20% according to various series,60,61 possibly higher (up to 47%) in the Japanese population.62 This variability can be ascribed in part to the different criteria adopted for the biochemical diagnosis of subclinical Cushing’s and in part to the variable degree of cortisol excess; this condition is by no means homogeneous. Usually, two or more of the following are considered indicative of subclinical Cushing’s syndrome: (1) incomplete cortisol suppression after low-dose dexamethasone; (2) low baseline ACTH levels; (3) absent cortisol circadian rhythm; (4) blunted ACTH response to CRH; (5) elevated UFC; (6) decreased DHEAS.63 Iodocholesterol scintigraphy may also be useful because exclusive uptake by the adrenal mass is indicative of excessive autonomous cortisol secretion with functional inhibition of the contralateral gland.64 On the other hand, the degree of uptake does not differentiate between overt and subclinical Cushing’s syndrome. Patients diagnosed with subclinical Cushing’s are somewhat older (mostly older than 50) than those with Cushing’s syndrome due to benign cortisol-secreting tumors, and the risk of developing overt Cushing’s syndrome in the short term appears low.46,65 However, even the mild excess in cortisol secretion observed in these patients is deleterious for metabolic, cardiovascular, and bone physiology, and removal of the “pretoxic” adrenal gland is followed in a number of cases by amelioration of diabetes, hypertension, insulin resistance, obesity, and parameters of bone turnover.66 A note of caution is necessary upon removal of the lesion: Suppression of the contralateral gland may lead to postsurgical adrenal insufficiency.65
Subclinical Cushing’s syndrome may also be the initial manifestation of AIMAH, and in this case, preclinical Cushing’s syndrome is the more correct term. In these patients, biochemical derangements and signs and symptoms of hypercortisolism may be subtle and wax and wane over prolonged periods of time. The involvement of both adrenal glands results in bilateral iodocholesterol uptake, although the timing of hyperplasia may be asynchronous (see AIMAH).
Imaging
Radiologic procedures for visualization of adrenal lesions comprise abdominal x-ray, sonography, computed tomography (CT), and magnetic resonance imaging (MRI), while iodocholesterol scintigraphy and the recently developed adrenal-specific positron emission tomography (PET) provide functional assessment of adrenal masses. Additional procedures such as arteriography and CT-guided biopsy are only rarely used in patients with adrenal Cushing’s syndrome (for detailed discussion see Chapter 105).
The aim of adrenal radiology in patients with adrenal Cushing’s syndrome is to visualize the lesion, define its morphologic features and eventual extension into neighboring structures and provide insights into the nature and function of the tissue. In addition, unrelated adrenal lesions such as silent pheochromocytoma, metastases, collision tumors, and even accessory spleens must be recognized.67,68 As with adrenal pathology, the results of individual imaging studies may be unequivocal and allow a straightforward diagnosis, whereas in other cases, CT, MRI, and scintigraphy may all be necessary. Unenhanced CT is usually the first-line procedure and may provide sufficient information to diagnose a benign lesion. Should malignancy be suspected, chemical-shift MRI may be performed to confirm the diagnosis and allow the staging of the lesion.
Computed Tomography
This technique provided a breakthrough in adrenal radiology, enabling easy visualization of the glands, given their location, morphology, and composition. Indeed, retroperitoneal fat provides an ideal background for recognition of the Y-shaped hyperlucent adrenal gland. The adrenal rim is usually straight or slightly concave, and any convexity should increase suspicion for underlying lesions. Adrenal size measurements are of little use because they largely depend on gland orientation.69
Adrenocortical adenomas, carcinomas, and macronodular hyperplasia are easily identified at unenhanced, thin-section (3 to 5 mm) CT scanning. As to their differentiation, adenomas usually present as a small (<3 cm), homogeneous, round mass with smooth margins and thin peripheral capsule (Fig. 102-9, left panel). Given the high steroid content of clear and compact cells, density of the mass is typically low, near that of water, and unenhanced attenuation values below 10 HU are commonly considered indicative of adenoma.19,70 Black adenomas present somewhat higher density than clear-cell yellow adenomas.71 Injection of a contrast medium, when performed, leads to homogeneous enhancement of the entire lesion, followed by rapid washout; attenuation values after enhancement do not allow clear distinction from nonadenomatous masses, whereas washout of contrast medium provides useful information for the differentiation from malignant lesions such as metastases.70 The unaffected gland is usually normal sized or occasionally smaller.15
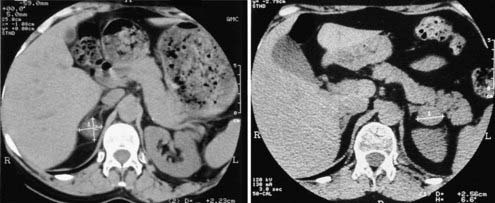
FIGURE 102-9. CT scan of cortisol-secreting adrenal adenoma (left panel) and adrenal nodular hyperplasia (right panel). This latter patient presented with a left-sided macronodule and bilateral enlarged glands.
Adrenocortical carcinomas commonly appear as large (>6 cm), irregularly shaped, inhomogeneous lesions with soft tissue density (>20 HU) and patchy enhancement. The borders of the lesion are often “smudged” and only rarely sharp. The heterogeneity of the mass is due to intratumoral necrosis, cystic degeneration, or calcification.19,69 These latter features, while extremely frequent in adrenal cancer, may be encountered also in degenerated adenomas72 and are not absolute criteria. Conversely, findings diagnostic for carcinoma are local spread (e.g., extension into the inferior vena cava, lymph node enlargement) or metastases. The size of the lesion, initially considered a useful criterion for the distinction of benign from malignant lesions, has increasingly been questioned; both large adenomas (>6 cm) and small carcinomas (<3 cm) are not infrequent.24 In addition, both CT and MRI may underestimate the size of the lesion by as much as 20%.73
In patients with AIMAH, CT scan most commonly reveals prominent glands with or without multiple large nodules. Massive macronodular hyperplasia is usually easily recognized because the glands appear markedly hypertrophic and multiple macronodules (>1 cm) can be seen in both adrenals14 (see Fig. 102-9, right panel). In other cases, only one gland appears to be affected.74 Conversely, in micronodular adrenal hyperplasia, the glands appear only slightly enlarged or even indistinguishable from normal. Indeed, a normal adrenal scan in a patient with ACTH-independent Cushing’s syndrome may provide indirect evidence of AIMAH.75
Adrenal radiology in patients with PPNAD varies from a normal appearance, slight enlargement of one or both adrenal glands, to unilateral or bilateral micronodules.12,76,77 Macronodules are more rarely encountered and mostly in patients older than 25 years of age.77 One feature typical to PPNAD is the “knobbly” and irregular outline of the glands, most likely due to internodular atrophy, resulting in a “beads-on-string” appearance.19,77
Magnetic Resonance Imaging
MRI easily visualizes both normal adrenal glands and adrenal masses by virtue of the abundant hydrogen ions in lipid tissues. In addition to two-dimensional imaging, which may reveal features typical of benign or malignant adrenocortical tumors (see previous paragraph), MRI allows the assessment of its lipid content and soft-tissue characterization via study of T2 signal intensity and relaxation times, dynamic perfusion studies, or chemical-shift opposed-phase images. Adrenocortical adenomas are isointense and slightly hyperintense relative to the liver on T1- and T2-weighted sequences, respectively,78,79 whereas carcinomas may display very high signal intensity on T2-weighted images.77,79 After gadolinium, carcinomas show marked enhancement and slow washout, in contrast with the rapid washout of benign lesions.79 Chemical-shift imaging reveals loss of signal in out-of-phase images in lipid-rich tissues and is used to differentiate adenomas from metastases80 but does not reliably distinguish between adenoma and carcinoma, since both usually display a loss of signal intensity.78 On balance, although some features may be highly indicative, the accuracy of MRI in differentiating adenoma from carcinoma is not absolute,16,67 and routine MRI is not recommended for patients with adrenal Cushing’s syndrome.19,79 In patients with adrenal carcinoma, however, MRI enables visualization of the mass along sagittal and coronal planes, which allows the definition of anatomic boundaries, compression of surrounding organs, and extent of caval invasion.24 MRI is the procedure of choice for Cushing’s syndrome during pregnancy81 and in childhood, because of safety concerns and because it allows visualization of the glands even in absence of retroperitoneal fat. The drawback of MRI in pediatric patients is the requirement for absolute standstill, which may be difficult to achieve.
Adrenal Scintigraphy
The advent of CT has shifted adrenal scintigraphy from the center stage of adrenal imaging to an adjunctive, complementary procedure. In the past, evaluation of adrenal uptake was an integral part of the diagnostic workup, providing information also for the differential diagnosis between ACTH-dependent and ACTH-independent Cushing’s syndrome. Nowadays it is mostly reserved for differentiation of adenoma from hyperplasia and for those rare cases of ectopic adrenal tissue or adrenal remnants after adrenalectomy. Adrenal scintigraphy relies on the ability of the adrenal cortex to capture and accumulate radiolabeled cholesterol. Tracers are 131I-19 iodocholesterol or its derivative 131I-6β-iodomethyl-19 norcholesterol (NP59), the latter offering sharper adrenal images owing to a fivefold greater adrenal concentration,82 and 75Se-6β-selenomethyl-19 norcholesterol—used mostly in Europe. For scintigraphy with 131I, adequate preparation with Lugol iodine drops or potassium iodide and laxatives reduces thyroid uptake and intestinal background. Scans can be obtained as early as 48 hours after tracer injection, but usable images are registered up to 7 to 10 days after administration.
Adrenal scintigraphy provides quantitative and qualitative data on adrenal function. In patients with benign adrenal hypercortisolism, radiolabeled cholesterol uptake may be increased compared to normal subjects, usually 0.7% of the administered dose compared to 0.3% in normal subjects83,84 (Fig. 102-10). This percentage, although indicative of adrenal hyperfunction, does not provide absolute certainty of excessive corticosteroid production and, indeed, does not appear to be directly correlated with indices of adrenal function in these patients.83 Conversely, qualitative data is of great help for localizing steroid-secreting lesions. Scintigraphy may yield several uptake patterns, as proposed by Gross83 (Table 102-1).
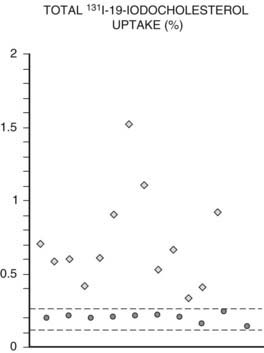
FIGURE 102-10. 131I-19-iodocholesterol total adrenal uptake in normal subjects (•) and patients with Cushing’s syndrome ( ). Dotted lines depict mean ±2 SD.
). Dotted lines depict mean ±2 SD.
(Adapted from Ortega D, Foz M, Doménech-Torné FM, Tresánchez JM: The diagnosis of Cushing’s syndrome using 131I-19-iodocholesterol uptake and adrenal imaging, Clin Endocrinol 13:148, 1980.)
Table 102-1. Uptake Patterns at Adrenal Scintigraphy
| Exclusive | Uptake on the side of the lesion, with nonvisualization of the contralateral gland |
| Prevalent | Asymmetric uptake by the tumor, with visualization of the contralateral gland |
| Symmetric | Comparable uptake by both glands |
| Discordant | Reduced or absent uptake on the side of the mass |
| Bilateral nonvisualization | No uptake on either side |
Exclusive or prevalent uptake on the side of the mass are considered concordant with the mass and confirm its functional activity as a steroid hormone–producing tissue and are taken to indicate a benign adrenocortical adenoma74,85 (Fig. 102-11A). Inhibition of ACTH secretion by autonomous cortisol production prevents the imaging of the contralateral gland. False-negative results have occasionally been reported with black adenomas, possibly a consequence of the prevalence of lipid-poor compact cells.86 In adrenocortical carcinoma, bilateral nonvisualization is the rule because the carcinoma itself does not accumulate tracer sufficiently, possibly a consequence of deranged steroidogenesis, and the contralateral gland is inhibited.85,87 However, some well-differentiated adrenocortical carcinomas and, rarely, also high-grade malignant carcinomas may accumulate the tracer, yielding exclusive or prevalent images.87 Malignant collision tumors may also display concordant uptake because of residual compressed cortical parenchyma within the nonadrenal neoplasm.59,88 It follows, therefore, that while bilateral nonvisualization is highly indicative of a malignant mass, concordant uptake does not uniformly represent benign disease. On the other hand, in patients with carcinoma concentrating the tracer, scintigraphy may enable the visualization of unsuspected metastases, thus providing invaluable preoperative information.89
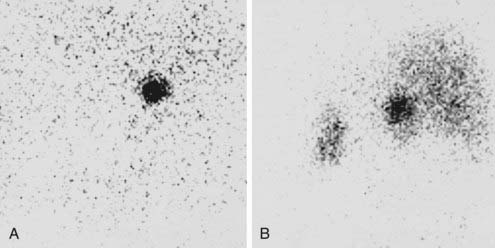
FIGURE 102-11. Scintigraphy in two patients with ACTH-independent Cushing’s syndrome due to primary adrenal disease. Images are posterior views obtained 1 week after 131I-19-norcholesterol injection. A, Exclusive uptake by a right-sided adrenocortical adenoma. B, Asymmetric uptake in a patient with macronodular adrenocortical hyperplasia.
(Courtesy Dr. Eugenio Reschini, Ospedale Maggiore, Milan, Italy.)
In patients with ACTH-independent Cushing’s syndrome and apparently normal adrenal radiology, scintigraphy becomes paramount in order to localize the lesion and establish the etiologic diagnosis.85,90 In those rare cases of adrenal adenoma not identified by CT or MRI, exclusive uptake will reveal the side of the lesion and direct the surgeon.90 In addition, scintigraphy may identify ectopic adenomas or lesions not recognized as adrenals at standard imaging.91 Bilateral symmetric or slightly asymmetric uptake characterizes adrenal hyperplasia (see Fig. 102-11B) and PPNAD,12,74,85,92 and adrenal scintigraphy may be the sole procedure to correctly diagnose these clinical entities. Both AIMAH and PPNAD may in fact present with normal or slightly enlarged glands or unilateral or bilateral nodules and need to be differentiated from adrenal adenoma.74,90,93 However, scintigraphy itself is not foolproof. Bilateral nonvisualization has been reported in patients with hyperplasia or dysplasia,15,93 as has unilateral uptake in a patient with PPNAD.12,94
Lastly, scintigraphy may be employed in recurrence of adrenal Cushing’s syndrome or Cushing’s disease after adrenalectomy to localize hyperfunctioning adrenal remnants.82,93
Adrenal Venous Sampling
Adrenal venous sampling, performed only in experienced centers, is profitably used to lateralize the source of aldosterone hypersecretion (see Chapter 102) but has been little used in adrenal Cushing’s syndrome. It might prove an interesting alternative to adrenal scintigraphy for those patients with bilateral adrenal masses in whom unilateral involvement is suspected95; large-scale studies are needed to validate its usefulness in adrenal Cushing’s syndrome.
Positron Emission Tomography
Whole-body positron emission tomography (PET) has been used in patients with adrenocortical carcinoma as an adjunctive procedure to detect and monitor metastases. Tracers employed so far are 18fluorine-fluorodeoxyglucose (FDG), the classical marker of enhanced glycolysis in malignant tumors, and 11carbon etomidate or metomidate, two inhibitors of 11β-hydroxylase. FDG-PET proved useful for the identification of neoplastic spread both prior to surgery and during follow-up, with markedly increased uptake detected in the site of the neoplasm and in metastases.96,97 Its use for the distinction between carcinoma and adenoma has been confirmed in larger series,98 although adrenocortical adenomas and hyperplasias may occasionally yield positive scans.98 PET using adrenal-specific tracers such as carbon-labeled etomidate or metomidate has yielded promising results for the detection of adrenocortical tumors and their metastases because uptake is clearly increased in adrenal cancer and derived lesions. The aim of this procedure, in contrast to 18F-FDG PET, is to establish the adrenocortical origin of the adrenal lesion; pheochromocytomas, metastases, and nonadrenal masses yield negative scans.99 Currently, only 11carbon-labeled metomidate is available, but 18fluorine tracers are being developed, and these may allow a more widespread use of adrenal-specific tracers in the future.
Interpretation of Adrenal Radiology
Adrenal radiology in patients with adrenal Cushing’s syndrome may reveal the presence of normal glands, unilateral or bilateral masses, or uniformly enlarged adrenals. A normal CT scan virtually excludes the presence of adenomatous or malignant lesions and can be taken to indicate micronodular hyperplasia or nodular dysplasia, the latter being more probable in young patients. Unilateral masses need to be categorized as malignant or benign, and this distinction remains most fraught in cases with intermediate radiologic features, since no procedure is decisive. Benign unilateral masses are most likely adrenocortical adenomas, although dominant nodules in AIMAH and even nodular dysplasia cannot be entirely excluded, and adrenal scintigraphy may be needed to discriminate between unilateral or bilateral disease. On the other hand, hypersecreting and nonhypersecreting adrenocortical adenomas present roughly the same CT/MRI features and cannot be distinguished by radiology.19,74 Bilateral masses with benign appearance are in all likelihood due to adrenal macronodular hyperplasia and, to a lesser extent, PPNAD. However, the possibility of bilateral carcinoma or adenoma, although exceptional, should not be forgotten. Lastly, bilaterally enlarged glands can be encountered both in adrenal hyperplasia and adrenal dysplasia.
Pathology of Cortisol-Secreting Adrenal Lesions
Adrenal lesions responsible for Cushing’s syndrome arise from the zona fasciculata, the middle layer of the adrenal cortex. The adrenal glands are located in the retroperitoneum, immediately above and in front of the kidneys. The mean weight of the adrenal gland is 5 to 6 g, length is approximately 4 cm, width 2.5 cm, and thickness 4 to 6 mm. The left adrenal is usually larger than the right gland and is generally placed more caudally. Men present slightly larger adrenal glands than women. On visual examination, the adrenal gland appears yellowish and triangular or semilunar in shape. The gland is surrounded by abundant retroperitoneal fat and closely wrapped in a thin fibrous capsule transversed by numerous vessels and fibrous branches. In addition to left and right adrenal glands, accessory adrenals may be hidden within the retroperitoneal connective tissue or inside distant organs such as the gonads. The adrenal cortex represents the external portion of the gland between the capsule and the internal adrenal medulla. The glandular epithelium of the cortex is organized into three different zones, each characterized by a different arrangement of polyhedral epithelial cells. The zona fasciculata, the most prominent layer, is composed of large lipid-rich cells, the so-called clear cells, arranged in radial columns. Clear cells present abundant agranular endoplasmic reticulum and well-developed mitochondria, two organelles necessary to steroid hormone production. The outer layer, the zona glomerulosa, is composed of highly granular cells arranged in alveolar structures, whereas the inner layer, the zona reticularis, is characterized by irregular nests of compact cells, poor in lipids and rich in lipofuscin granules. The adrenal cortex is extensively innervated by the autonomous nervous system, and nerve terminals abut directly on cortical cells, thereby influencing cortical cell function.
Four main adrenal lesions may be detected in patients with adrenal Cushing’s syndrome: a nodule (either isolated or within hyperplastic adrenals), adenoma, carcinoma, and nodular dysplasia. Distinction of hyperplastic nodules from adenoma, as well as adenoma from carcinoma, requires careful pathologic examination not only of the nodule itself but also of the surrounding adrenal tissue (Table 102-2). Indeed, different pathologic systems are used for the diagnosis of these lesions; each offers different advantages and is burdened by different drawbacks (see Chapter 106).
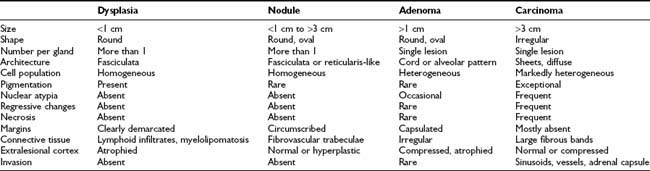
Hyperplastic nodules are usually distinguished from adenomas by the following features: less than 1 cm in diameter, oval or round shape, homogeneous clear-cell population organized in zona fasciculata– or zona reticularis–like architecture, rare smooth cell nests or cordlike structures, with well developed fibrovascular trabeculae.100–102 Nodules are usually more than one per gland, circumscribed but not encapsulated, and adjacent tissue is normal or hyperplastic. Conversely, adenomas are round or oval lesions of more than 1 cm in diameter, with heterogeneous cell population comprising both clear and compact cells in irregular cord or alveolar patterns. The adenoma is usually a single, encapsulated lesion surrounded by atrophic and compressed cortex. Both hyperplastic nodules and adenomatous lesions are surrounded by a rich network of sinusoidal vessels and dilated intercellular spaces and present well-developed, variably sized and shaped mitochondria and prominent smooth endoplasmic reticulum.103 Hyperplastic nodules and adenomas present as yellow, lipid-rich lesions, but occasionally lesions are composed of lipofuscin-containing compact cells with abundant eosinophilic cytoplasm, resembling zona reticularis cells and giving rise to black nodules or the so-called black adenoma.71,102 In patients with AIMAH, adrenal pathology depends on the progression and the extent of the lesions. Most patients present with huge adrenals (up to 500 g) in which the normal architecture is subverted by the presence of big, multiple, yellow or black nodules, some as large as 7 cm, surrounded by internodular hyperplasia (macronodular hyperplasia). Occasionally, the adrenals may be only slightly enlarged, with multiple small nodules (micronodular hyperplasia) or only one gland might be affected, presenting one or multiple nodules (unilateral hyperplasia). Intriguingly, hyperplastic nodules and adenomas have been reported in the same adrenal gland of patients with Cushing’s syndrome,102 as have bilateral adenomas or multiple adenomas in the same gland.104,105 In these cases, nodules or adenomas may present different cell composition (e.g., clear or compact cells) and secretory status (hypersecreting or nonhypersecreting) and affect the two glands at different time points. Indeed, bilateral adenomas as well as hyperplastic nodules have been removed several years apart.106,107
A unique variant of bilateral nodular involvement is PPNAD, a condition in which adrenals are small to normal-sized and occupied by multiple small, dark nodules (from 1 to 2 mm to 3 cm in diameter) containing large, heavily pigmented, granular eosinophilic cells (rarely clear cells). Pigmented granules contain lipofuscin and neuromelanin.26,108 Nodules are sharply demarcated, although not encapsulated, and surrounded by atrophic cortical tissue—this provides the major distinction from bilateral hyperplasia. The lesions are usually positioned deep within the adrenal cortex, nearly straddling the corticomedullary junction, whereas the remaining cortex may lose the normal zonation pattern12 (Fig. 102-12). Some nodules may show lymphocytic infiltration (mostly T-helper cells) within and without the nodule, as well as myelolipomatosis.109,110 As occurs for cortical tumors, positive immunostaining for neuroendocrine markers, such as synaptophysin and neuron-specific enolase, may be detected.111 Occasionally, nodularity may be asymmetric and present as a unilateral mass, or more rarely, nodules are markedly prominent, and adrenals appear enlarged,77 mimicking adrenal macronodular hyperplasia.
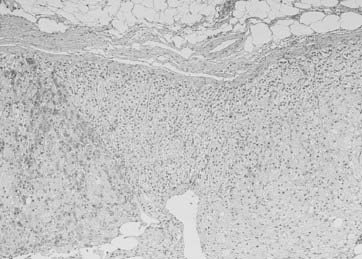
FIGURE 102-12. Pathologic findings in PPNAD. The two nodules are separated by internodular atrophic cortex. Note absence of capsule around nodules and foci of myelolipomatosis.
(Courtesy Dr. Aidan Carney, Mayo Clinic, Rochester, MN.)
The diagnosis of adrenal carcinoma is straightforward if the neoplasm presents typical malignant features (“anaplastic carcinoma”) such as necrosis, hemorrhage, abundant mitoses, and nuclear atypia, together with local or hematogenous spread. In most cases, however, Cushing’s syndrome is associated with “well-differentiated carcinoma,” because steroidogenesis proceeds inefficiently in anaplastic tumors, and these precursors are devoid of significant biological activity.112 The distinction of well-differentiated carcinoma from adrenocortical adenoma relies on tallies from different pathologic scoring systems, the most used being those of Weiss,113 Hough,114 and van Slooten.115 These diagnostic algorithms are based upon histologic features such as necrosis, architectural changes, nuclear atypia, hyperchromasia, mitotic activity and atypical mitosis, nucleolar appearance, cytoplasmic vacuolization, cellular pleomorphism, fibrosis, and capsular and vascular invasion. The Hough scoring system also takes tumor weight and clinical/endocrine features into account (e.g., 17-ketosteroid excretion and response to ACTH, Cushing’s syndrome with or without virilization, body weight loss).114 While widely accepted, none of these systems offers complete diagnostic certainty,116 and the definite diagnosis of malignancy remains local invasion or metastasis. Borderline pathologic grading for benign lesions should lead to close follow-up because development of malignant tumors on the same site of a previously removed adenoma117,118 questions the initial diagnosis of “benign adrenocortical adenoma.” Ultrastructural analysis usually reveals lipid-depleted cells with altered mitochondrial and smooth endoplasmic reticulum architecture.103 Immunohistochemical studies evaluating p53 expression, MiB-1 or Ki-67 labeling, AgNOR counting, MHC class II antigen expression, and c-myc intracellular distribution revealed a differential pattern in benign and malignant lesions, but heterogeneity of neoplastic phenotypes and the resulting overlap between the two neoplasms precludes the identification of a secure profile of malignancy.119 Likewise, DNA ploidy analysis, comparative genomic hybridization, candidate gene mutations, or differential expression (most notably IGF-II; see Pathogenesis of Adrenal Tumors heading) revealed distinct differences between adenoma and carcinoma but, again, none appeared to ensure absolute diagnostic accuracy.119 Thus the current state of the art calls for prolonged follow-up also for apparently benign lesions for timely detection of signs of malignancy.
Several variants of adrenocortical tumors have been associated with Cushing’s syndrome, such as myxoid adrenocortical carcinoma, composed of polygonal basophilic cells arranged in arborizing cords surrounding myxoma-like large acellular spaces,120 or oncocytic adrenal tumors, usually benign nonfunctioning lesions composed of mitochondria-rich eosinophilic cells.121 Combined adrenal adenoma-myelolipoma characterized by intermingled cortical, bone marrow, and lipid cells has also been reported in conjunction with adrenal Cushing’s syndrome.122 Even more complex are mixed corticomedullary tumors, in which tumoral cortical cells are intimately admixed with pheochromocytes,123 or cortical tumors with neuroendocrine differentiation.124,125 In the latter case, malignant or benign adrenocortical cells present neuroendocrine features, such as dense core granules and positivity for synaptophysin and neurospecific enolase, and the tumor may give rise to excess secretion of various peptides, even ACTH itself.44 Pigmented adrenal lesions are usually benign (i.e., “black adenoma” or PPNAD), although a malignant pigmented lesion has also been reported.126 Finally, cortisol-secreting adenoma and carcinoma have been reported in the two adrenals of the same subject,127 as have bilateral carcinomas.128 On balance, tumors of the adrenal cortex usually display obvious benign or malignant features but occasionally may present with mixed forms or bizarre combinations that challenge the pathologist.
Pathogenesis of Adrenal Tumors
Mechanisms leading to adrenocortical tumorigenesis will be discussed in detail in Chapter 106; this issue will be reviewed briefly here, focusing on features of cortisol-secreting tumors.
Studies performed in the past decade suggest a common multistep process leading from polyclonal hyperplasia, to polyclonal or monoclonal adenomatous lesions, then to single-clone malignant cell expansion. This process, which is in keeping with Knudson’s two-hit hypothesis, is apparent in adrenal lesions at different stages,129,130 possibly even within the same adrenal.131 Further, genetic heterogeneity in terms of clonality, chromosomal alterations, and proliferation kinetics characterizes all three adrenal lesions, and no single feature allows the distinction between these forms. The common origin of adenoma and carcinoma is also supported by the fact that the same chromosomal regions appear to be affected in benign and malignant lesions.132 Loci of putative oncogenes (sites of chromosomal gain) and tumor-suppressor genes (sites of chromosomal loss) have been identified on chromosomes 17, 4, 5, and 9 and chromosomes 1, 2, and 11, respectively.133 Loss of heterozygosity is frequently detected on 11p15.5 (site of IGF2/H19/CDKN1C), 17p13 (site of TP53), 11q13 (site of MEN1), 13q (site of the retinoblastoma gene RB1), 9p (site of CDKN2A), and less frequently on 1p (site of neuroblastoma candidate gene NBL1) and 3p (site of the von Hippel-Lindau gene VHL).133,134 These changes, most notably the first two, are common in malignant adrenal tumors and are believed to represent pivotal events for adrenocortical tumorigenesis (see Chapter 106). Further, alterations in IGF-2 and p53 can be encountered in Beckwith-Wiedemann and Li-Fraumeni syndromes, respectively, two hereditary conditions presenting, among others, adrenocortical tumors. Germinal TP53 mutations are also linked to adrenal cancer predisposition in southern Brazil, a region with inexplicably high incidence of pediatric adrenal carcinoma.135 Mutation analysis of factors involved in tumorigenesis in other endocrine tissues (e.g., G proteins, ras) failed to identify significant alterations,119 nor did the analysis of cortex-specific genes, such as the 21-hydroxylase gene CYP21A2.136 The ACTH receptor appears unchanged in patients with adrenal disease,137 although a mutation in MC2R has been reported in one patient with adrenal hyperplasia138 (see heading ACTH-Independent Adrenal Hyperplasia). Of interest, somatic mutations of PRKAR1A, the gene involved in Carney’s complex, and loss of heterozygosity at its locus, 17q22-24, have been detected in adrenal tumors and adrenal hyperplasia,139,140 as well as alterations in protein kinase A subunits,141 suggesting an extensive involvement of the cAMP pathway in adrenocortical disease. Lastly, recent microarray studies opened several new avenues of research for adrenocortical lesions, most notably into the Wnt/β-catenin signaling pathway142 and the inhibin/activin complex.143
INHERITED FAMILIAL SYNDROMES
Adrenocortical tumors can be encountered in a number of genetic syndromes, such as multiple endocrine neoplasia (MEN) type 1, MAS, familial adenomatous polyposis and Carney’s complex.
Multiple Endocrine Neoplasia Type I
A link between MEN1 (Wermer’s syndrome) and adrenal tumors is suggested by the frequent loss of heterozygosity at 11q13, site of the menin gene, in adrenal cancers.133 However, no mutation in the MEN1 gene itself has been reported in cortisol-secreting sporadic adrenocortical tumors.144 Conversely, up to 70% of patients with MEN1 present adrenal lesions, mostly nodular and diffuse hyperplasia, adenomas, or rarely, carcinomas.145 Cushing’s syndrome in the context of MEN1, however, is mostly associated with pituitary ACTH-secreting adenoma, since adrenal lesions are only rarely accompanied by hypercortisolism.146
McCune-Albright Syndrome
Since the first description of bony lesions, skin pigmentation, and precocious puberty by Albright in 1937, the spectrum of endocrine abnormalities associated with this syndrome has progressively increased to include thyroid, parathyroid, pituitary, and adrenal disorders. Adrenal Cushing’s syndrome develops in less than 10% of patients with MAS, usually in the first year after birth, possibly even before sexual precocity and fibrous dysplasia.17 Adrenal histology usually reveals bilateral nodular hyperplasia or, rarely, adenoma; interestingly, these lesions have also been detected at autopsy in patients who did not present Cushingoid signs during their lifetime. MAS is known to be due to early postzygotic mutations of the stimulatory guanine nucleotide-binding protein (Gsα; GNAS1), which can be detected in multiple tissues, including the adrenal gland,147 and also in patients with MAS and adrenal Cushing’s syndrome described so far.17,148 Conversely, GNAS1 mutations are exceptional in patients with sporadic cortisol-secreting adrenal adenomas.149 GNAS1 mutations have been reported in adult patients with Cushing’s syndrome due to bilateral adrenal hyperplasia without other features of MAS,150,151 suggesting that mutational Gsα activation could be involved in adrenal hyperplasia.
Familial Adenomatous Polyposis
Patients affected by this autosomal dominant inherited disorder develop extensive adenomatous polyps of the colon and a variety of extracolonic manifestations comprising desmoid tumors, osteomas, pigmented retinal lesions, and adrenocortical neoplasms. Adrenocortical lesions consist in adenomas, bilateral nodular hyperplasia, or rarely, carcinomas and are mostly nonhypersecreting.152 Cushing’s syndrome is a rare occurrence both in classic familial adenomatous polyposis152 and its variant, Gardner’s syndrome.153 Familial adenomatous polyposis has been causally linked to germline mutations of the adenomatous polyposis coli (APC) gene located on chromosome 5, usually inactivating mutations accompanied by loss of the normal allele. It is worth recalling that inactivation of the APC gene leads to degradation of β-catenin, a recent putative player in adrenal tumorigenesis.142 Somatic mutations of APC have been detected in a wide variety of other human carcinomas but none so far in sporadic adrenocortical tumors.
Carney’s Complex
Carney’s complex represents the only etiology of Cushing’s syndrome in which a genetic defect has been identified. Indeed, intensive work by two groups of researchers led to the short-listing of two probable loci, on the short arm of chromosome 2 and on 17q24, followed by the identification of inactivating mutations in the protein kinase A regulatory subunit type 1α.154,155 Carney’s complex is discussed in greater detail later (see heading Primary Pigmented Nodular Adrenal Dysplasia).
STEROIDOGENIC ENZYMES IN ADRENAL LESIONS
Several studies have evaluated steroidogenesis in adrenal Cushing’s syndrome (see Figure in Chapter 96). Cortisol concentration in tumoral specimens is 2- to 50-fold greater in adenomas than in normal glands,156 attesting to the greater secretory output. Steroidogenesis usually proceeds without hindrance to its principal end product, cortisol, but individual steroidogenic enzymes may become rate-limiting in hypersecreting adrenals,157 the extent and involvement of individual enzymes being quite variable. Analysis of steroidogenic enzymes yielded somewhat conflicting results, with increased 21-hydroxylase and 17α-hydroxylase synthesis and activity in cortisol-secreting adenomas as the most constant finding.112,156,158 Increased 17,20-lyase activity reportedly occurs in adenomas producing androgens in addition to cortisol, possibly a consequence of increased cytochrome b5 expression.159 A relative deficiency of 11β-hydroxylase has been suggested by some authors,160 although not confirmed by others.158 Conversely, 3β-hydroxysteroid dehydrogenase and cholesterol desmolase appear substantially unchanged in cortisol-secreting adenomas.112,158 In adrenal carcinoma, cortisol secretion per gram of tumoral tissue is mostly reduced,160 as are individual steroidogenic enzymes.28,112,161 Of interest, malignant cells may fail to synthesize the full complement of steroidogenic enzymes,28,161 thus possibly explaining ineffective steroidogenesis. Shift from excess aldosterone to excess cortisol production has also been reported in malignant tumors, associated with changes in steroidogenic enzyme synthesis and attesting to the plasticity of tumoral tissues.162 The low efficiency of cortisol secretion is also a feature of macronodular hyperplasia,100 and variable expression of steroidogenic enzymes in individual cortical cells has been observed.163 In contrast, nodules from patients with PPNAD all present intense immunostaining for steroidogenic enzymes.163 Lastly, low levels of expression and activity of 11β-hydroxysteroid dehydrogenase type 2, the enzyme which inactivates cortisol, have been detected in adenomatous cortisol-secreting adrenals and may represent an additional modulator of total cortisol levels.164,165
REGULATION OF STEROID SYNTHESIS IN ADRENAL LESIONS
In adrenal Cushing’s syndrome, cortisol secretion is by definition ACTH-independent. However, a considerable proportion of adenomas, and to a lesser extent carcinomas, maintain ACTH-responsiveness in vivo and in vitro,7,166 and the relevance of this preserved ACTH sensitivity in tumors which secrete cortisol autonomously remains unclear. The ACTH receptor (also known as the melanocortin 2 receptor, MC2-R) is expressed in the vast majority of cortisol-secreting adenomas, even overexpressed compared with the normal adrenal cortex, according to some authors,166,167 whereas cortisol-secreting carcinomas present low levels of MC2R mRNA, possibly a consequence of dedifferentiation and loss of heterozygosity at this locus.167 ACTH is known to up-regulate its own receptor at low concentrations and exert the opposite effect at higher doses,168 but no direct association between ACTH receptor mRNA and ACTH plasma levels has been detected in various forms of Cushing’s syndrome.169 Stimulation of steroid secretion in adrenocortical tumor cells occurs through activation of protein kinase C, together with the main protein kinase A–cAMP pathway.170 Of interest, factors other than ACTH may also increase cAMP levels in malignant but not in normal adrenal cells, as has been demonstrated for catecholamines and thyrotropin,171 indicating a loss of specificity of the transductional machinery in tumoral tissues (see heading ACTH-Independent Adrenal Hyperplasia). Moreover, ACTH is also an indirect adrenocortical mitogen acting through factors such as basic fibroblast growth factor (bFGF), insulin-like growth factor 2 (IGF-2), epidermal growth factor (EGF), and transforming growth factor (TGF) α or β.172,173 These mediators are responsible for the growth-promoting effect of ACTH in vivo, whereas in vitro, ACTH exerts a direct antimitogenic activity.172 Similarly divergent are the effects of N-terminal pro-opiomelanocortin (POMC), which appears to stimulate mitogenic activity via the secretory serine protease (AsP) pathway in vitro174 but is devoid of mitogenic effects in Pomc-null mice in vivo.175
One brief mention is warranted for the regulation of cortisol synthesis by corticosteroids themselves. Evidence for this ultra-short negative feedback has been garnered by in vivo and in vitro studies, although paradoxically, a stimulatory, permissive effect of cortisol or dexamethasone on ACTH-stimulated cortisol secretion has been reported by some authors.176 The direct action of corticosteroids on steroidogenesis is presumably mediated by glucocorticoid receptors themselves, since both binding and induction of gene synthesis have been demonstrated in the adrenal cortex.167,177 Expression of the glucocorticoid receptor is apparently reduced in cortisol-secreting tumors.167,178
ACTH-Independent Adrenal Hyperplasia
ACTH-independent adrenal hyperplasia was recognized as a cause of Cushing’s syndrome following the description of several case reports which defied classification.179,180 Considerable confusion arose initially from the fact that some features of ACTH-dependent adrenal hyperplasia (i.e., Cushing’s disease) overlapped with ACTH-independent adrenal hyperplasia; indeed, “macronodular Cushing’s syndrome” once comprised both entities. Advances in pathology,100 as well as the identification of factors other than ACTH capable of modulating cortisol hypersecretion, enabled a more precise definition and classification.14,181,182 In view of its most common presentation, we will refer to this entity with the currently used acronym, AIMAH: ACTH-independent macronodular adrenal hyperplasia.
Currently accepted criteria for the diagnosis of AIMAH are the presence of bilaterally enlarged adrenals containing one or more nonpigmented nodules and the presence of suppressed and/or unresponsive ACTH levels.14,100 Internodular tissue was initially described as atrophic100 but may also be normal or even hyperplastic, thus does not represent a distinctive element.14,182 On average, glands weigh five times normal (e.g., 20 to 120 g) and present large, well-circumscribed nodules grossly distorting adrenal cortex architecture (see heading Pathology of Cortisol-Secreting Adrenal Lesions). The size of nodules usually ranges from 0.5 cm to 2 to 3 cm, although exceptionally large lesions, up to 10 cm in diameter, have been described.183 Nodules are composed of lipid-laden clear cells in cord or nest-like structures interspersed by small islands of lipid-poor compact cells,100,184 foci of angiomyelolipomatosis and lymphocytic infiltrates. Cellular and nuclear pleomorphism are usually absent. Unilateral macronodular hyperplasia, as well as diffuse bilateral hyperplasia, have also been described in patients with ACTH-independent hypercortisolism,14,185 possibly representing different points in the continuum of AIMAH.
The large size of adrenals is in marked contrast with the often mild clinical picture, corroborating in vitro evidence of inefficient steroidogenesis by cortical cells.100,163,184 Indeed, cytochrome P450 expression and activity are reduced compared to that of cells derived from adrenal adenoma100,184; thus the adrenal mass may have to attain a considerable volume for hypercortisolism to become manifest. Subclinical Cushing’s syndrome is increasingly reported in patients with ACTH-independent adrenal hyperplasia,186 occasionally evolving with progressive adrenal enlargement and development of frank hypercortisolism.187,188
Salient features of hypercortisolism due to AIMAH are later age at diagnosis (usually around 50 years of age), the even gender distribution, and an unusually long time to diagnosis, on average 4 years from the appearance of the first symptoms, compared to 2 years for cortisol-secreting adenomas.14,182 One patient developed full-blown hypercortisolism 7 years after the incidental detection of bilateral adrenal masses.188 The youngest and oldest reported patients with ACTH-independent adrenal hyperplasia were 17 and 74 years old, respectively. In patients with aberrant receptor-induced hyperplasia (see later), hypercortisolism may follow the course of ligand levels, as exemplified by the course of LH-dependent Cushing’s syndrome or by low morning serum cortisol levels in the fasted patient with food-dependent Cushing’s syndrome.
By definition, ACTH levels are suppressed in patients with ACTH-independent adrenal hyperplasia, but the detection of low-normal ACTH concentrations does not exclude the diagnosis.14,182 As in other causes of adrenal Cushing’s syndrome, CRH testing provides proof of ACTH independency. Inferior petrosal sinus sampling should not be performed, because significant post-CRH center-periphery ACTH gradients have been reported in several patients, presumably reflecting incomplete suppression of pituitary corticotrophs by mild or episodic hypercortisolism.189 Prior to the availability of ACTH assays, adrenal autonomy from ACTH was established by means of absent dexamethasone suppression or metyrapone stimulation of adrenal steroids. These tests, however, do not guarantee complete diagnostic accuracy; indeed, the number of patients with Cushing’s disease and adrenal nodules who fail to suppress after dexamethasone is not negligible,190 and specificity of metyrapone stimulation is known to be suboptimal.47 The distinction between ACTH-independent adrenal hyperplasia and unilateral adrenal lesions or PPNAD relies on imaging, both morphologic and functional, and pathology (see earlier discussion).
Major diagnostic difficulties may be encountered in the distinction from longstanding Cushing’s disease with ACTH levels in the lower third of the normal range and from adrenal adenoma or even carcinoma, if only one large dominant nodule is visible at adrenal imaging. The distinction is obviously essential to a correct surgical approach.
PATHOGENESIS
ACTH-independent adrenal hyperplasia presents a heterogeneous etiology, comprising legitimate and illegitimate adrenal growth factors and gene mutations. In one of the first descriptions of ACTH-independent adrenal hyperplasia, proliferation of small cortical cells with maturational arrest or incomplete maturation was hypothesized to cause hyperplasia.100 Causes identified so far, however, account for only part of reported patients, and other mechanisms are likely to be involved. Indeed, microarray studies in AIMAH tissues identified a host of potential candidate genes,191,192 in part overlapping with genes involved in adrenal tumorigenesis, such as the Wnt signaling pathway, and cytogenetic analysis revealed frequent allelic losses in 2p16 and 17q22, two sites linked to PPNAD,140 in adrenal hyperplastic tissues.
Legitimate Adrenal Growth Factor
Adrenal hyperplasia was initially believed to result from prolonged stimulation by ACTH followed by autonomization of adrenal secretion. Several case reports describing transition from ACTH-dependent to ACTH-independent hypercortisolism supported this hypothesis.193,194 In fact, ACTH induces exaggerated cortisol increases in patients with adrenal hyperplasia, both in vivo and in vitro, attesting to an exquisite adrenal sensitivity to ACTH, in contrast with the barely preserved responsiveness of adrenal adenomas and carcinomas. Indeed, the cortisol response to cosyntropin represents the standard against which responses to other agents are compared (see later). This pathogenetic hypothesis, while theoretically still tenable, has been superseded by subsequent developments.
A role for ACTH cannot be excluded, since locally produced ACTH participates in the intraadrenal CRH/ACTH modulation of corticosteroid release and adrenal proliferation.195 In two patients with adrenal hyperplasia, cortisol secretion was driven by intraadrenal ACTH production. In one patient, ACTH was produced by an adrenocortical adenoma presenting neuroendocrine features,196 whereas in the other patient, steroidogenic cells with positivity for Leydig-cell markers were the site of ACTH synthesis.197 Interestingly, plasma ACTH levels were high in the former but not in the latter patient, and the diagnostic workup comprising adrenal vein sampling was indicative of ectopic ACTH secretion.196 Altered sensitivity to the adrenal ACTH receptor might be an alternative mechanism. Indeed, a homozygous MC2R germline mutation was identified in a patient with Cushing’s syndrome and ACTH-independent adrenal hyperplasia; functional studies revealed constitutive activation of the F278C mutated receptor.138 This case is unique; no mutations in MC2R were detected in other patients with ACTH-independent adrenal hyperplasia.151,198 Quantitation of MC2R expression in adrenals from patients responsive to illicit receptor stimulants yielded contrasting results, with some studies indicative of increased expression,199 others observing reduced expression,192,200 and others again observing expression comparable to normal adrenocortical tissues.201 Variability in clinical and molecular phenotypes might explain these differences.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


