Fig. 18.1
Maturation of the antigen-presenting DCs by an adjuvant. Myeloid cells appear to represent the response of innate immunity. Pattern molecules of microbes and denatured self cells behaving like non-self material, activate DCs, and tissue-resident macrophages. IFNs and cytokines are liberated from these cells in response to adjuvant. In addition, activation of DCs result in enhancement of the expression of the co-stimulators and major histocompatibility complex class I, leading to activation of a cellular effectors (NK, CD4 T cells, CTL, etc.). Here we especially outline the roles of the TLR2–MyD88 and TLR3–TICAM-1 pathways in activation of the cellular immunity followed by activation of innate immunity by the adjuvant for cancer immunotherapy
18.3 Bacterial Adjuvants for Therapeutics Revisited
The phenomenon of infection-inducing tumor regression was known experientially in the eighteenth century. William Coley performed a trial in which bacteria was injected into humans (cancer patients) with the goal of treatment in the 1880s, when the mechanism of understanding lymphocyte antigen recognition had not yet been defined. In 1891, the therapeutic results of the bacterial therapy (mixed bacillus of Streptococcus pyogenes and Serratia marcescens) for cancer patients was published (Coley 1891). In today’s terms, it is so-called ‘adjuvant immunotherapy’, although problems, such as TAAs being dependent on the patient (van der Bruggen et al. 1991), were undetermined. This method prescribed simultaneous administration of bacterial foreign antigens and PAMP, in contemporary terms, to the cancer patients, and was activating acquired immunity from innate immunity. Although anti-tumor CTL induction was not proven, it was effective in some cases. However, there were severe side effects and cases of death. Since the Federal Institute for Drugs and Medical Devices of Germany was not able to reproduce this vaccine’s validity, it was withdrawn in 1990. In the future, attenuating bacteria incorporating the genes of TAAs will be a device that is sufficient as a therapeutic.
Since the 1960s in Japan, cancer immunotherapy has been developed using BCG of the tubercular vaccine (Yasumoto et al. 1979; Kodama et al. 2009). These examples are BCG-CWS by Yamamura and Azuma (Azuma et al. 1974) and Maruyama vaccine (Maruyama 1971), both of which were crude extracts of BCG bacillus. There are case reports of effective monotherapy use of BCG-CWS administration by Kodama and coworkers at the Osaka Medical Center for Cancer (Kodama et al. 2009). The BCG treatment raised the rate of complete remission to 70 % or more in a bladder cancer (transitional–epithelial cancer) as a standard treatment (Lamm et al. 1991). BCG-CWS was reported as an agonist of TLR2/4 by Tsuji et al. in 1999 (Tsuji et al. 2000). BCG-CWS consists of mycolic acid moieties with TDM, arabinogalactan, and peptidoglycan (containing MDP) (Seya et al. 2002). Ishikawa et al. and Schoenen et al. have reported that TDM is a ligand for a C-type lectin receptor, Mincle (Ishikawa et al. 2009; Schoenen et al. 2010). The peptidoglycan portion contains MDP, which is reported to be the active center for TLR2 agonistic activity, but, unexpectedly, synthesized MDP failed to contribute to TLR2 agonistic activity (Uehori et al. 2005). The MDP portion therefore will not act as a center of the activity of BCG-CWS. MDP will hardly confer effective anticancer activity to the patients unless the other region of PGN is attached. To date, the whole PGN has been impossible to synthesize chemically. Sumitomo Pharmaceuticals recently provided highly refined BCG-CWS under GMP regulation and called SMP105, which showed anticancer activity in mouse tumor-implant models (Murata 2008). There was, however, inconsistency, and tumor regression depended upon MyD88 but was independent of TLR2. These results suggest that multifarious or complicated immune responses induced by the mixture of BCG origin were more advantageous to the anticancer immune effect than the single isolated or synthesized compound. The reader is referred to the literature for further details of the mechanism of activation of TLR2 in the immune system by PAMP for immunotherapy (Vacchelli et al. 2013).
18.4 Viral Adjuvants for Therapeutics Revisited
It has been known for a long time that viral infection affects the prognosis of patients with cancer. Since discovery of IFN (later defined as type I IFN) in 1957, study of this mechanism has been underway (Taniguchi et al. 1980). From the 1960s, Levy and others started anti-tumor immunotherapy that prescribes a virus product (dsRNA analog polyI:C) as an inducing factor of IFN for treatment with tumor-implant mice (Levy et al. 1969). PolyI:C has been used since the 1970s for patients with cancer. In 1979, IFN-β complementary DNA was cloned and applied to cancer patients for the purpose of direct immuno-cytokine therapy (Levine et al. 1979). Although these reagents contributed to tumor regression in a number of cancers in humans, severe side effects were exhibited (fever, arthralgia, myalgia, neurological manifestation, endotoxin-like shock, etc.). Therefore, it is not yet clinically applied, except for in some restricted types of cancers (metastatic renal cancer, hairy cell leukemia). Another problem was its expense.
By that time, it had become clear that IFN is expressed by stimulation with dsRNA of the virus. Nevertheless, polyI:C as a dsRNA analog was used because the sequence-specific synthesis of long RNA was impossible throughout the studies of dsRNA medication. Indeed, polyI:C was greeted at the beginning in the 1970s with great expectation as a promising medicine including cancer immunotherapy. However, it being stated as “untolerable” (proof of being impossible to administer) increased as a clinical trial progressed. In order to reduce the side effects of polyI:C, Levy and coworkers mixed the RNase inhibitor LC (polyL-Lysine and methylcellulose) into polyI:C, and used it in a clinical trial. However, the maximal therapeutic dose was still very low, and patients died before an effective amount was determined. An example of the severe side effects was renal failure causing death at a dose of 27 mg/m2, which represented a sign of cytokine storm. PolyI:CLC (Hiltonol®) and polyI:C12U (Ampligen®) are still used for immunotherapy. The cytoplasmic RIG-I/MDA5 pathway may be activated by these reagents, although both are provisionally called TLR3 agonists.
Although IFN-inducing activity was proved in human volunteers using 1.6 mg subcutaneous administration, in this low dose cellular effectors appear to be hardly guided (Caskey et al. 2011). The phase II examination was still ongoing in 2012.
18.5 Innate Immune Adjuvants Revisited
Although Coley and Levy started their anticancer treatments wholly independently, both reached the TLR adjuvant therapy by PAMP. Moreover, both obtained suggestions from the observation of infection-mediated tumor regression and developed adjuvant therapies for cancer by artificial administration of PAMP to patients. Because adjuvants were regarded as evil principles in the cancer therapy field as compared with surgery, radiation, and chemotherapy, common agreement to this therapy could not be reached easily. Yet, scientific understanding and reconsideration of cancer immunity are called for nowadays based on the fact that eligible patients are extremely restricted by the sole peptide vaccine therapy. As the 2000s have progressed, an understanding of the importance of an adjuvant has gradually permeated in association with the knowledge of DC maturation in the field of cancer immunotherapy (Fig. 18.1). If we made an effort to faithfully reproduce the results of the clinical study of Coley and Levy as science, the adjuvant must have been effective against cancer. In accordance with this view, the practical research regarding TLR2 and TLR3 as adjuvants (for human use) is discussed below.
18.6 TLR2 as an Adjuvant Receptor
The development of crystal structure analysis made it clear that TLR generally formed a dimer to recognize specific PAMPs. TLR2 discriminates and recognizes a detailed difference of PAMP structure by heterodimerization (Kang et al. 2009). TLR2/TLR6 recognize Pam2 lipopeptides and TLR2/TLR1 recognize Pam3 lipopeptides (Fig. 18.2). Seemingly, PGN will be recognized by the TLR2 dimer, although it remains unproven since chemical synthesis of PGN is unfeasible.
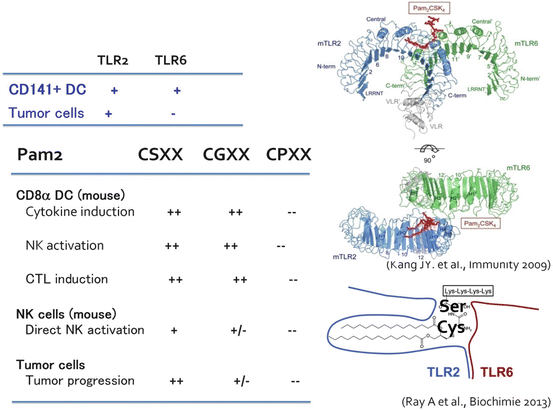
Fig. 18.2
The crystal structure of TLR2 and its ligand recognition. Kang et al. (2009) first submitted the crystal structures of TLR2/TLR6 and TLR2/TLR1 in the presence of binding ligands. Taken together with the result of cristalized protein analysis (Ray et al. 2013), the amino acid sequences of the peptide portion of conventional TLR2 lipopeptide will regulate the response of the macrophage in a tumor (TLR2-positive), and CD141+ DCs (TLR2-/TLR6-positive). Development of TLR2 adjuvant which does not have tumor proliferation activity may be feasible based on this concept
Pam2 and Pam3 differentially activate TLR2 signaling, which should reflect the difference in the TLR2 signal caused by PGN according to the distinct TLR complex. Pam2 lipopeptides have various peptide sequences depending upon bacterial species, and bacteria express various Pam2 lipopeptides on the cell membrane. This will be one of the reasons why activation of TLR2 changes with bacterial species. Although the diversity of the lipopeptide sequences in which amino acids from the second amino acid (the first is always Cys) differ among bacterial species is natural providence, it will be a problem of the Pam2/3 adjuvants if those differences have any physiological implication (Azuma et al. 2010).
Unexpected findings were obtained when we compared the functions of DC TLR2 with tumor-infiltrating macrophage TLR2 (Maruyama et al. 2015). NK activation and IL-10 secretion are dependent by DC TLR2 on a peptide sequence of the Pam2 lipopeptide (Azuma et al. 2010; Yamazaki et al. 2011). TLR2 in tumor-resident macrophages is engaged in tumor progression and metastasis (Kim et al. 2009). Macrophages in tumors are a unique target for therapy. That is, unlike TLR2 of other healthy cells, the TLR2/TLR6 pocket for antigen-presenting DCs recognizes Pam2-Cys-Ser-X-X and Pam2-Cys-Gly-X-X, and induces cytokine production and NK activation (Fig. 18.2), but the activity disappeared in Cys-Pro and Cys-Leu substitution (Azuma et al. 2010). On the other hand, the peptide sequence recognized by TLR2 of tumor cells and NK cells is only Pam2-Cys-Ser-X-X. In the case of Pam2 lipopeptides, NK activation usually initiates cell damage other than IFN-γ and CD69 upregulation by NK cells (Azuma et al. 2010). Type I IFN is never induced by TLR2/MyD88 signal.
If Pam2 lipopeptides with a peptide sequence of the DC type (Cys-Gly) are used for medication, based on the above knowledge, this TLR2 agonist can selectively activate DCs and then cellular immunity without involving tumor implication (Takeda et al. 2015). In the case of tumor-associated macrophage-dependent ligands, such as versican, TLR2 exerts protumor activity (Kim et al. 2009). DC-type Pam2-Cys-Gly-X-X simultaneously cross-primes CTL in a MyD88-dependent manner (Takeda et al. 2015). Although Pam2/3 lipopeptides allow myeloid cells to liberate a lot of cytokines, NK cell activation ability is not as strong in these TLR2 agonists as in TLR3 agonists, which should be attributable to the functional properties of Pam2 that block NK cell activation (Yamazaki et al. 2012). DCs mature via the p38 MAPK pathway in this context through activation of AP1 transcription family proteins (Takeda et al. 2015). Future research is needed to determine what kind of molecular mechanism lies in the MyD88 downstream that participates in DC maturation and CTL cross-priming.
18.7 TLR3 as an Adjuvant Receptor
It has become clear that TLR3 forms dimers from crystal structure analysis, and recognizes dsRNA in a dimeric form (Bell et al. 2006). The extracellular domain (consisting of LRRs (Leucine rich repeat)) of TLR3 recognizes a dsRNA structure at two places, and even if the other spacer RNA part is an imperfect stem (with many mismatches), TLR3 recognizes dsRNA without any functional problem (Tatematsu et al. 2013). The structure of the 5′/3′ end of RNA does not participate in TLR3’s RNA recognition either. That is, TLR3 recognizes the dsRNA structural motif (Fig. 18.3), which is not recognized by RIG-I/MDA5. Based on these background data (Matsumoto and Seya 2008), it is expected that a TLR3-specific ligand will not have cytokine toxicity, which is totally due to RIG-I/MDA5 signaling, in systemic administration (Seya et al. 2013). A design completed from in vitro synthetic compounds of RNA and the TLR3-specific agonist will be perfect for chemical synthesis (Matsumoto et al. 2015).
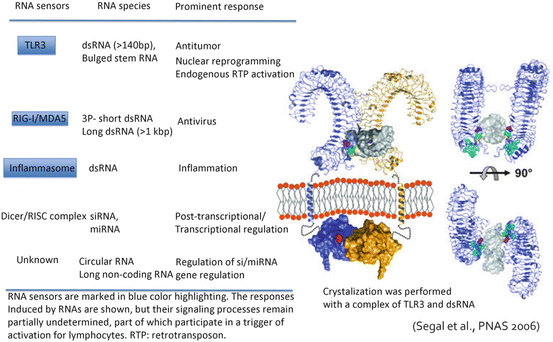
Fig. 18.3
The crystal structure of TLR3 and its ligand recognition. The crystal structure of the complex of TLR3 and dsRNA was presented by Liu et al. 2008. The figure shows the dsRNA-binding site on the C terminus region of the extracellular TLR3 protein. Another dsRNA-binding site is situated in the N terminus of TLR3. Neither dimer formation nor TICAM-1 signal transmission is spoiled, although DC TLR3 receives enzymatic processing in the endosome. The physiological significance of the limited proteolysis of TLR3 by endosomal cathepsins remains unknown. Although the TLR3 pathway in DCs performs strong driving of cellular effectors, other unresolved activities remain to be defined, such as promotion of nuclear reprogramming and activation of endogenous retrotransposon in epithelial cells and fibroblasts, which was described recently (left figure). Here we compare these reported RNA responses of TLR3 with those of other RNA sensors
Systemic administration of polyI:C resulted in cytokinemia in mice bearing implant tumors. In those models, however, TLR3 plays a pivotal role in tumor regression. PolyI:C activates NK cells (Akazawa et al. 2007), the mechanism of which is thought to be by induction of INAM via the IRF-3 and IFNAR1 pathways (Ebihara et al. 2010; Kasamatsu et al. 2014). Importantly, early induction of IRF-3 activation by polyI:C is indispensable for introduction of CD8α DC (mouse) and CD141–DC-mediated cross-presentation (Azuma et al. 2012, 2015; Kasamatsu et al. 2015). Strikingly, polyI:C converts tumor-supporting macrophages to tumor-suppressing ones (Shime et al. 2012, 2014). In addition, polyI:C expresses direct anti-tumor activity by inducing apoptosis/necroptosis to some types of tumors (Paone et al. 2008; Conforti et al. 2010; Seya et al. 2012; Takemura et al. 2015).
If systemic administration of this TLR3-agonistic RNA is carried out and only TLR3 response is reproduced in humans, the importance of TLR3 in the evoking of cellular immune response by RNA can be proved (Galluzzi et al. 2012). Although we have reproduced this theory in mice, a future study is needed in humans (since GMP material is underdeveloped). The reason that old clinical trials of polyI:C were intolerable to patients is thought to be the toxicity by high cytokinemia. Since TLR3 is expressed only in myeloid and epithelial cells (Matsumoto et al. 2003), TLR3 barely causes systemic IFN induction like RIG-I/MDA5. The lessons from the nature of such TLR3 agonists may enable us to develop a new therapeutics involving NK/CTL inducers with few side effects for humans (Fig. 18.3). That is, the cellular immune effecters can be induced by a synthetic RNA in vivo, independent of cytokinemia (Matsumoto et al. 2015). Anti-tumor immunotherapy will be attained by extensive administration of this TLR3 agonist.
Related posts:
 A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
A New Mode of Cytotoxic T Lymphocyte Induction Secondary to Alteration of the Peptide/Major Histocompatibility Complex Class I Repertoire by Antigen Processing Defects
In Vivo Targeting of Dendritic Cells with Artificial Adjuvant Vector Cells (aAVC) as a Novel Cancer Immunotherapy
 PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
 Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
Anchorage-Dependent Multicellular Aggregate Formation Induces CD44 High Cancer Stem Cell-Like Phenotypes in Adult T Cell Leukemia/Lymphoma Cells
 Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
Enhancement of Efficacy of Wilms’ Tumor Gene WT1 Product-derived Peptide Cancer Vaccine by Co-administration with Immunopotentiating Agents: Lessons from Mouse Models
 Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Regulation of Antigen Presentation by Dendritic Cells and Its Application to Cancer Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


