Darier’s sign.
Blood tests carried out show a normal full blood count, liver function tests, and bone profile (calcium and vitamin D level).
Is a skin biopsy necessary to confirm the diagnosis of CM?
A. No
B. Yes
Answer: A, No.
UP is the most common presentation of mastocytosis in children and adults (Figures 12.2 and 12.3). In children it usually develops in the first year of life and can fully resolve without treatment. In adults, the mean or median age of onset for UP is between 20 and 40 years.
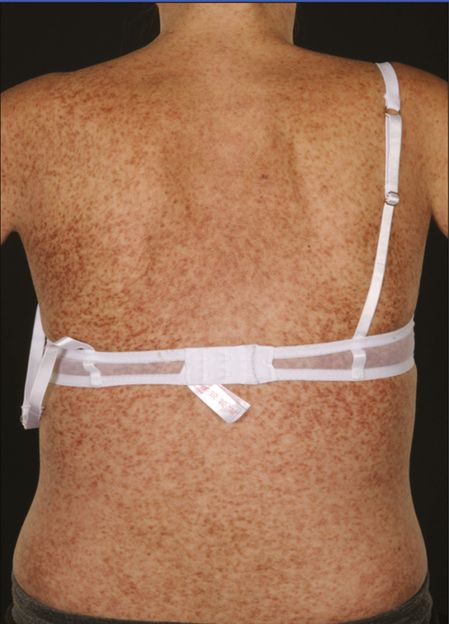
Diffuse cutaneous urticaria pigmentosa.
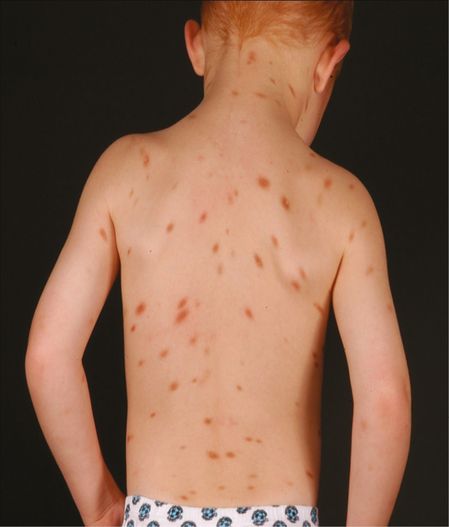
Cutaneous mastocytosis (urticaria pigmentosa).
Other dermatological presentations of a mast cell disorder include telangiectasia macularis eruptive perstans, which develops as persistent red macules with or without obvious telangiectasia. These lesions are not Darier’s-positive.
A very rare form of CM usually presenting in the neonatal period is diffuse CM, where there are no discrete lesions and the skin appears thickened and doughy but is not pigmented. Trauma or scratching can lead to blister formation. Children with this presentation are at a higher risk of systemic symptoms, including anaphylaxis and diarrhea. This often resolves spontaneously. Solitary mastocytomas can also be seen in infants as isolated lesions up to 3–4 cm in diameter (Figure 12.4) and nearly all involute over the first few years of childhood.
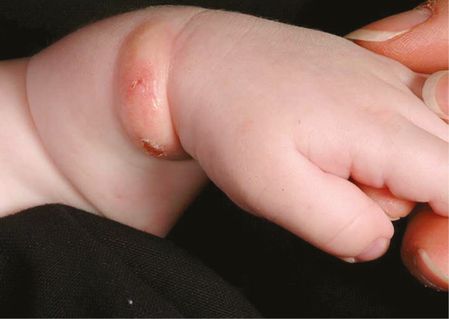
Solitary mastocytoma.
Patients with a classical presentation of UP who are relatively asymptomatic may not require a skin biopsy. However, it is recommended one be performed to confirm the diagnosis if in doubt or if there is an atypical presentation. In general, skin biopsy is avoided in young children and infants to prevent scarring.
Case 1: part 2
This woman’s tryptase result is 28 ng/mL. Would that be an indication to carry out a bone marrow biopsy?
A. No
B. Yes
Answer: B, Yes.
A serum tryptase level of > 20 ng/mL is one of the minor criteria in the WHO classification to diagnose SM (Table 12.2).2
| Systemic mastocytosis is defined by finding one major + one minor, or three minor criteria on bone marrow biopsy |
| 1. Major: multifocal aggregates of at least 15 mast cells in bone marrow or other organ (not skin) (Figures 12.5, 12.6, 12.7) |
| 2. Minor: |
| (a) >25% of mast cells in infiltrates are spindle-shaped in bone marrow biopsy or other tissue, or >25% of mast cells in bone marrow aspirate smears are immature or atypical (Figure 12.8) |
| (b) Activating mutations in KIT (e.g., codon 816) in bone marrow, blood, or other tissue (not skin) |
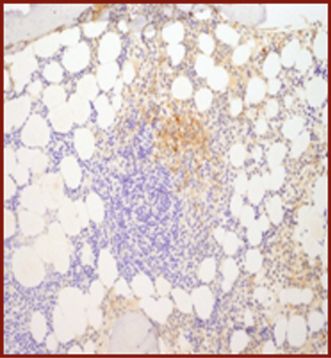
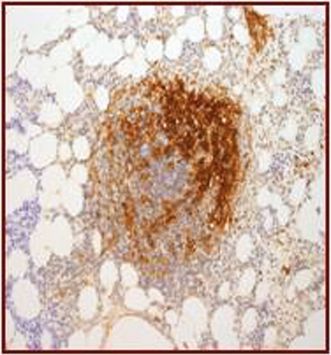
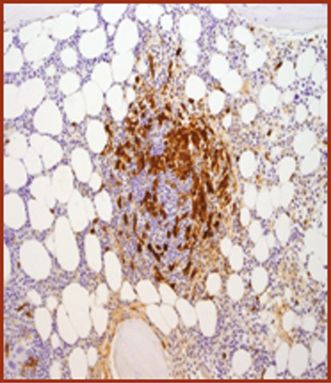
| (c) Co-expression of CD117 with CD2 and/or CD25 in bone marrow mast cells, blood, or other tissue (not skin) (Figures 12.9, 12.10, 12.11, 12.12) |
| (d) Serum tryptase >20 μg/L (unless associated clonal myeloid disorder) |
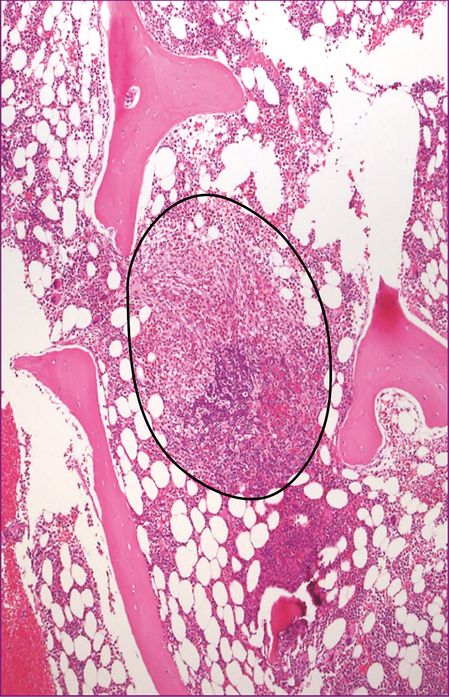
Bone marrow trephine – major criteria/basic lesion.
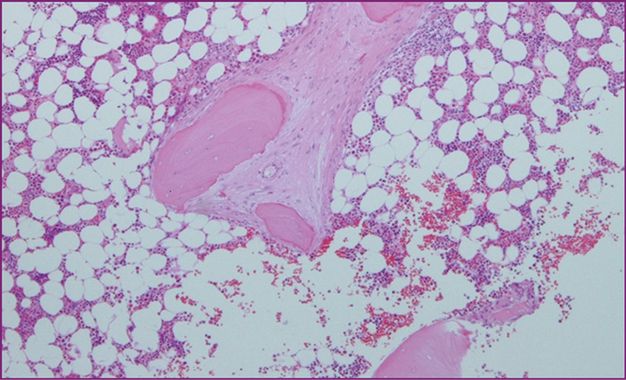
Peritrabecular infiltrate of mast cells with fibrosis.
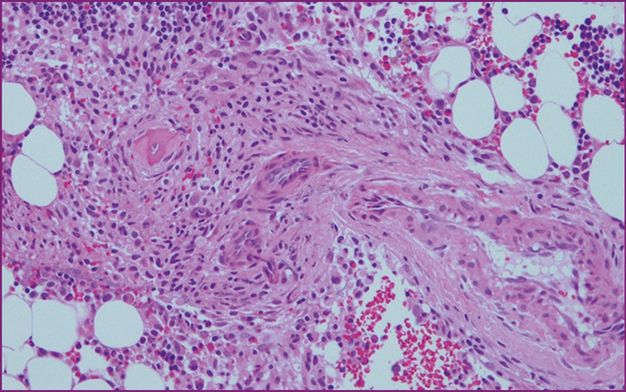
Perivascular infiltrate of mast cells.
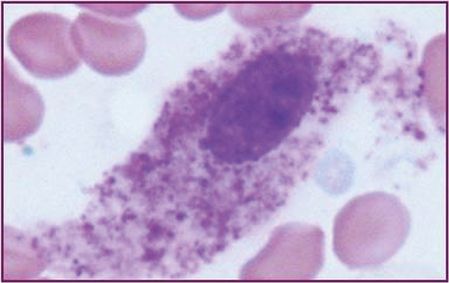
Bone marrow aspirate ×100 (oil).
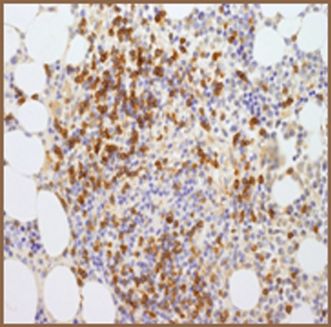
CD117.
CD2.
CD25.
Mast cell tryptase.
It is important to bear in mind that tryptase levels are variable and will be elevated in a patient who is affected by mediator symptoms, e.g., flushing, pruritus, or gastrointestinal symptoms.3 In addition, a persistently raised tryptase may not be diagnostic of SM and rarely may be raised in other conditions, e.g., chronic myeloid leukemia, myelodysplastic syndromes, atopic eczema, chronic urticaria, or renal failure.4 Basal tryptase levels of >20 ng/mL on at least two occasions should be used as a threshold to discuss a bone marrow in adult patients. Some patients may have indolent SM (ISM) with a tryptase level <20 ng/mL and the rationale for whether a bone marrow is needed will need to be decided on an individual clinical basis.1,5 Patients with unexplained hypotensive/syncopal episodes, unexplained anaphylaxis, anaphylaxis associated with Hymenoptra venom, or unexplained osteoporosis with an elevated serum tryptase levels >15 ng/mL should be considered for a bone marrow biopsy to exclude SM.6 Patients with organomegaly and abnormal blood counts need diagnostic bone marrows regardless of the tryptase levels to exclude an associated hematological non-mast cell disorder (AHNMD).
Serum tryptase is the most useful screening test (basal and during an episode related to mast cell activation) and will guide further diagnostic tests, including a bone marrow biopsy. Other diagnostic tests such as 24-hour urine methylimidazole acetic acid, prostaglandin D2 and 11-β-prostaglandin F2α levels can help, but are not universally available.
Case 1: part 3
A bone marrow biopsy is carried out, and this demonstrates the presence of clonal mast cells (MCT+, CD117+,CD25+,CD2–, and CD30+). A mutation analysis is carried out. What is the most common mutation found in patients with SM?
A. FIP1L1-PDGRFA
B. KIT D816L
C. KIT V506G
D. KIT D816V
E. KIT F522C
Answer: B, KIT D816V.
Somatic gain-of-function mutations in the KIT tyrosine kinase domain have been described in up to 90–95% of adult patients with SM. This leads to constitutive autophosphorylation of the receptor, resulting in autonomous differentiation and survival of mast cells. The most common mutation is the D816V point mutation (WHO minor criterion for the diagnosis of SM) and was identified in 1995.7 Rarer somatic KIT mutations have been described in less than 5% of patients with SM and include V506G, D815K, D816Y, ins VI815-816, D816F, D816H, and D820G. The small minority of patients who are D816V-negative will be sensitive to imatinib therapy if needed.
It is important to evaluate the patient’s full blood count and bone marrow aspirate for eosinophilia and to exclude the presence of the FIP1L1-PDGRFA as a small subset of patients have SM with hypereosinophilic syndrome and are sensitive to treatment with imatinib.
Familial mastocytosis is rare and is associated with rare germline KIT mutations such as F522C and A533D.8,9
Case 1: summary and management
This patient has a diagnosis of ISM, presenting with UP and mild mediator symptoms.
Treatment will depend on the severity of the itching and also how much the cosmetic appearance of the rash affects her. Empiric symptomatic treatment options include antihistamines for pruritus (anti-H1 and anti-H2 antihistamines) starting with chlorpheniramine, loratidine (non-sedating), cetirizine, rupatidine10 and fexofenadine. Occasionally doses much higher than those recommended in the formulary may be needed by some patients or several combinations of antihistamines to achieve symptom relief. Application of potent topical steroids under polyethene occlusion for 2 weeks may be of benefit for some patients, but this needs to be carried out under the care of a dermatologist to minimize the risks of side effects and to monitor the outcome.
Cosmetic therapy is limited to phototherapy with ultraviolet (UV) A, narrow-band UVA or UVA with psoralen. These modalities are most effective in patients who have cutaneous lesions as well as symptoms including pruritus and flushing, but there are no reports of complete cosmetic resolution of lesions. These may fade or lighten in appearance and this may be transient. Case reports of the disappearance of skin lesions with the use of cytoreductive treatments such as midostaurin, tyrosine kinase inhibitors, and cladribine (2CdA) have been reported.11,12 However, the potential risk of cytotoxicity outweighs the benefit in patients with CM or ISM.
Up to 30% of patients with mastocytosis can have osteoporosis regardless of the classification.13 The mechanism of this is thought to be as a result of bone remodeling influenced by mast cell mediator activity in the bone marrow matrix involving interleukin-6 and receptor nuclear factor kappa-B ligand (RANKL). Baseline screening for osteopenia/osteoporosis (dual-energy X-ray absorptiometry (DEXA) scan) is recommended in addition to skeletal survey to assess for the presence of lytic lesions if patients are symptomatic with bone pain (“C” finding in the WHO criteria).
It is recommended that all patients with SM, regardless of a previous history of anaphylaxis, be advised to carry an Epi-Pen (epinephrine autoinjector) as there is an increased risk of anaphylaxis in patients with mastocytosis. The reported prevalence of anaphylaxis is approximately 30–50%14 compared to 0.05–0.2% in the normal population.15
Case 2: part 1
A 52-year-old man was referred to the hematologist for an opinion.
He presented with abdominal symptoms over the preceding 6–8 months: loose motions with a frequency of 6–8× daily, abdominal fullness, and discomfort associated with cramps.
On examination he was noted to have hepatomegaly (3 cm below the costal margin and splenomegaly 4 cm below the costal margin). He had no palpable lymphadenopathy or skin lesions.
His full blood count showed a hemoglobin of 118 g/L, white cell count 12.5 × 109/L, neutrophils 7.0 × 109/L, lymphocytes 3.0 × 109/L, eosinophils 1.5 × 109/L, monocytes 0.8 × 109/L, basophils 0.2 × 109/L, and platelet count 125 × 109/L. His liver function tests were normal, as were his calcium and vitamin D levels. His tryptase level was 225 ng/mL.
His bone marrow investigations confirmed the presence of spindle-shaped mast cells with a 40% mast cell bulk in his trephine which expressed CD117, CD25, MCT, weak CD2, and CD30. His aspirate showed mild dyserythropoiesis: 6% mast cells (Figure 12.10) with no other associated hematological disorder. Cytogenetics were normal and his mutation status was D816V-positive.
What is his diagnosis?
A. ISM
B. Smoldering SM
C. Aggressive SM
D. SM with an AHNMD
E. Mast cell leukemia
Answer: B, Smoldering SM.
Confirmation of SM is commonly by bone marrow biopsy/tissue biopsy, demonstrating an aberrant mast cell immunophenotype with clonal mast cells expressing mutations in the encoding receptor for stem cell factor KIT.16
This patient has organomegaly with mild anemia and no biochemical evidence of end-organ damage. His “B” findings include a mast cell disease burden of >30% and serum tryptase >200 ng/mL and so he would fall into the subcategory of smoldering SM. Patients with smoldering SM make up approximately 5% of patients diagnosed with SM and the natural progression of the disease is unknown, as there are few epidemiological data.
Case 2: part 2
Would you treat this patient with cytoreductive treatment?
A. Yes
B. No
Answer: B, No.
Close monitoring of this patient is advised but no cytoreductive treatment at this point in time. Some patients with smoldering SM remain stable and may not progress to end-organ failure or aggressive SM. This particular patient has gastrointestinal symptoms and these are likely to be due to mediator release. Treatment would be symptomatic with anti-H1 and anti-H2 antihistamines. Sodium cromoglicate, a mast cell stabilizer, can be particularly effective in the management of bowel symptoms. A starting dose of 200 mg bd and then titrating an increasing dose according to symptoms whilst monitoring the liver profile is useful. It is important to note that some patients with SM require doses that exceed the formulary-recommended doses to control their symptoms, i.e., up to 400 mg qds, or may need a combination of antihistamines and mast cell stabilizers. It would be prudent to carry out endoscopic investigations to exclude any alternative etiologies. Assessment for osteopenia/osteoporosis should be carried out and advice given about the risk of anaphylaxis.
Related posts:
 Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
Longstanding polycythemia vera and essential thrombocythemia: monitoring and management goals
 Incorporating symptomatic assessment in therapy choice for patients with myeloproliferative neoplasms
Incorporating symptomatic assessment in therapy choice for patients with myeloproliferative neoplasms
 Accurately assessing risk in your myeloproliferative neoplasm patient
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
Accurately assessing risk in your myeloproliferative neoplasm patient
Managing acute vascular events in patients with myeloproliferative neoplasms
Challenging thrombotic scenarios in the myeloproliferative neoplasms: splanchnic vein thrombosis and others
 Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Atypical chronic myeloid leukemia and chronic neutrophilic leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


