WHIPPLE’S DISEASE
MATTHIAS MAIWALD AND DAVID A. RELMAN
In a seminal autopsy report in 1907, George H. Whipple provided a thorough and articulate description of the disease that now bears his name (1). His publication illustrated nearly all the key features that define this clinical entity. However, there was no mention of neurologic manifestations, and no postmortem examination of the central nervous system (CNS). More than 50 years later, the incidence and significance of CNS involvement in Whipple’s disease began to be appreciated (2). We now know that Whipple’s disease is a systemic disorder that typically affects the gastrointestinal tract and that the CNS is one of the most common sites of extraintestinal involvement, along with the lymphatic system and the heart (3,4). In addition, the CNS is the most common site of relapse after antimicrobial treatment of non-CNS disease (5). Furthermore, it has been suggested that early CNS infection occurs in most or all patients with Whipple’s disease, although only a subset of patients develop symptomatic or radiologically apparent CNS disease (6).
Classical Whipple’s disease primarily affects middle-aged men and usually presents with arthralgias, abdominal pain, fever, diarrhea, malabsorption, and weight loss (4,7–12). A fastidious bacterium, Tropheryma whipplei, is the causal agent of the disease; it is an actinomycete (class Actinobacteria) on the basis of molecular phylogenetic analysis (13–16). Whipple’s disease can present with protean manifestations, especially in the setting of symptomatic CNS involvement. Thus, delays or failures in the diagnosis of this disease are common. The prognosis of CNS Whipple’s disease is generally serious and is worsened by delays in, or withholding of appropriate antimicrobial therapy (5,17).
This chapter focuses on the CNS aspects of Whipple’s disease. Relatively little attention is given to the systemic (non-CNS) features of this disorder. These have been well described in a number of other reviews (4,10,18–21). Because it is an extension of the CNS, a discussion of Whipple’s disease involving the eye is included (see the section “Clinical Manifestations”).
ETIOLOGY
George Whipple concluded from histology and special stains that the subject of his case report suffered from a disorder of lipid metabolism (1). Although he observed bacilliform structures in silver-stained sections of a lymph node, he did not interpret this finding as related to the pathogenesis of the disease. However, beginning in the 1950s and 1960s, evidence started to accumulate that Whipple’s disease is caused by a distinct species of bacterium. First, bacteria with a characteristic uniform morphology are consistently seen by electron microscopy in pathologically affected tissues (22–25). These organisms are usually numerous, are recognized and phagocytosed by tissue macrophages, and undergo binary fission in areas of pathology. Second, patients with clinical manifestations of Whipple’s disease respond to antibacterial therapy (4,26). Third, intact bacteria disappear with clinical response to antibiotics and reappear during clinical relapse (27). Fourth, specific bacterial DNA sequences from T. whipplei are consistently detected in and have been shown to hybridize to areas of pathology (14,28). Finally, after several decades of unsuccessful attempts, the bacterium was isolated in culture, first from a heart valve specimen (29) and subsequently from other clinical specimen types, including intestinal tissue (30) and cerebrospinal fluid (CSF) (31).
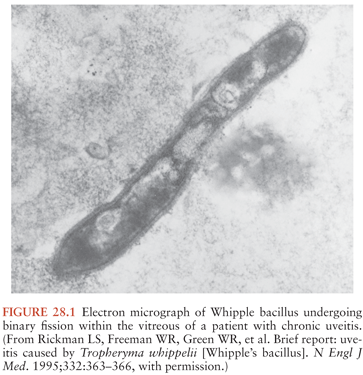
The Whipple’s disease bacterium (T. whipplei) is rod-shaped, measures about 0.2 µm by 1 to 2 µm, and is surrounded by a 20-nm-thick cell wall. The outermost layer surrounding the bacterium consists of a symmetric membrane that morphologically resembles those of eukaryotic origin (25). An inner electron-dense layer within the wall reacts strongly with periodic acid–Schiff (PAS) reagent and accounts for the PAS-positive staining pattern of the bacteria and their remnants within macrophage vacuoles. The organism stains weakly gram positive and is not acid fast. Examples of typical morphology, when viewed by electron microscopy, are shown in Figure 28.1 and in several references (24,25,27). Characterization of the bacterium at the molecular level was initially achieved by broad-range 16S ribosomal DNA (rDNA) polymerase chain reaction (PCR): a unique bacterial 16S ribosomal RNA (rRNA) gene sequence was found directly in affected tissues (13,14). Phylogenetic analysis indicated that the bacterium belongs to the actinomycetes (13,14); however, its relationships to the other known actinomycetes are relatively distant (15). The name Tropheryma whipplei (initially proposed as Tropheryma whippelii) is based on the Greek words trophe, nourishment, and eryma, barrier, because of the malabsorption in classical Whipple’s disease and to honor George Whipple as the discoverer of the illness (14,16,32).
Cultivation of T. whipplei in the laboratory was finally successful in prolonged co-culture with human fibroblast cells (29). In the initial culture setup, a bacterial generation time of about 18 days was observed (29), but subsequent reports estimated the doubling time at between 28 hours and 4 days (31,33,34). In any case, this is among the slowest growth rates ever reported for a medically relevant bacterium. Subsequently, a cell-free culture medium was designed, consisting of tissue culture medium supplemented with amino acids which the bacterium cannot synthesize on its own (34), and additional strains—both in cell culture and in cell-free medium—have been cultivated from various other clinical specimen types (35,36). Of relevance for the neurologic aspects of Whipple’s disease is the successful isolation of T. whipplei from CSF specimens; this includes CSF from neurologically symptomatic as well as asymptomatic patients and from a patient in the treatment-free period after prolonged antibiotic therapy (31,37). These findings indicate that T. whipplei is viable and presumably is able to replicate in the CNS. Nevertheless, at the present time, culture is not a suitable tool for routine diagnostic purposes in the workup of possible Whipple’s disease.
Two T. whipplei isolates from culture, one from CSF and another from an infected heart valve, served as the basis for genome sequencing projects (38,39). It is now established that the genome of T. whipplei is quite small, slightly less than 1 Mbp, and bears the features of a reduced-genome organism that is dependent on a host for synthesis of amino acids and for energy metabolism. At the same time, there is a relative abundance of predicted surface molecules, and this includes a unique protein family, termed the WiSP family (for T. whipplei surface proteins). The genome also possesses a number of “built-in” mechanisms to create genetic and antigenic variation, and these are predicted to generate considerable variability among the organism’s surface molecules. It is hypothesized that these features are linked to immune evasion and thus to the organism’s ability to sustain a chronic infection.
The natural occurrence and reservoirs of T. whipplei are a topic of ongoing research and debate. The first record of the bacterium’s occurrence outside diseased human tissues was provided by the detection of specific sequences in wastewater from sewage treatment plants (40), suggesting that T. whipplei may have an environmental reservoir and reside within polymicrobial communities. Several diagnostic PCR studies only rarely found positive results in pathologically unaffected human tissues (41–44). However, several other studies found T. whipplei–specific DNA sequences in saliva, gastric juice, intestinal biopsies, and stool of asymptomatic persons (45–49), prompting debate as to the reliability and generalizability of these findings (20,50). More recent studies have reported positivity rates of 2% to 4% in stool and 0.2% in saliva of healthy adults and 8% in stool and 2% in saliva of sewage workers, both in France, and the results were confirmed by a second PCR assay (48,49). Further evidence was provided with the detection of T. whipplei in stool and saliva of asymptomatic household members of patients with Whipple’s disease, including some of the same bacterial genotypes as in the respective patients (51). As a consequence, it has been postulated that there is an asymptomatic carrier state with T. whipplei and that the organism is a commensal bacterium of humans that causes Whipple’s disease only in a subset of predisposed individuals (20,36,50).
Differences between T. whipplei strains were first detected in the 16S-23S rRNA intergenic spacer sequence, and up to seven different spacer sequence types have been described (52–54). Subsequent analyses found several highly variable genomic sequences (HVGS) that formed the basis for an improved strain typing system (55); this genotyping system has been used in several epidemiologic investigations in France and in Senegal (51,56,57). However, although some geographical differences in strain distribution have been noticed, so far, no specific associations between different T. whipplei strains and particular disease manifestations or carrier states have been identified (36).
EPIDEMIOLOGY
Classical Whipple’s disease with intestinal involvement is a rare entity. It has been estimated that about 20 to 30 cases of Whipple’s disease are reported each year in the literature (4) and that the total number of published cases since Whipple’s original description is less than 2,000 (12). Available epidemiologic data indicate that the disease almost exclusively affects Caucasians, that the mean age at diagnosis is 50 to 60 years, that there is a strong proclivity for males, and that people in the farming trades and other outdoor professions are proportionately overrepresented (4,10–12,58). However, available incidence estimates almost certainly underrepresent the true number of cases and tend to exclude cases with nonclassical pathology or purely extraintestinal disease. In fact, recent reports indicate that the involvement of T. whipplei in culture-negative endocarditis without intestinal manifestations may be more common than would be expected from the incidence of classical Whipple’s disease, suggesting that endocarditis might follow a different epidemiologic pattern (59,60). Other reports implicate T. whipplei in acute transient illnesses, such as episodes of febrile illnesses in Senegal (61) and acute diarrhea in children in France (56), although the causative role of the organism in these instances is uncertain.
Epidemiologic assessment of CNS Whipple’s disease is hampered by frequent uncertainties surrounding the diagnosis of cases. Often, investigators diagnose CNS Whipple’s disease on the basis of CNS clinical manifestations plus confirmed diagnosis of Whipple’s disease pathology at other anatomic sites, typically in the small intestines or mesenteric lymph nodes. In cases with positive histopathology in the small intestine and confirmed by either electron microscopy or a well-validated PCR test, the diagnosis is firm (4,18,62). However, there is an increasing number of reports and case series of presumed, isolated CNS Whipple’s disease in the absence of other organ manifestations (63–66). In a considerable fraction of these published cases, the diagnosis has been made solely on the basis of positive PAS staining results and/or insufficiently validated PCR tests. As discussed in the section “Diagnosis and Differential Diagnosis” (later in this chapter), such diagnostic approaches lack the necessary specificity to be able to confirm CNS Whipple’s disease.
Current literature suggests that the demographics of CNS Whipple’s disease are roughly similar to those of Whipple’s disease cases involving all sites, although relevant information is limited. In the large review by Dobbins (4), among 28 patients with CNS Whipple’s disease for whom an age is listed, the mean is 40 years. A published series of 11 cases of CNS Whipple’s disease lists a 9:2 male:female ratio and a mean age at onset of 49 years (67). Another review of CNS Whipple’s disease (68) provides a mean age of 50 years and a male:female ratio of 8:2 among a group of 70 patients with predominant but not exclusive neurologic presentation, and a mean age of 42 years and a male:female ratio of about 1:1 among a group of 21 patients with presumed isolated neurologic manifestations.
Among several published case series of systemic and/or classical intestinal Whipple’s disease, the proportion of patients with neurologic manifestations ranges between slightly less than 10% and slightly more than 40% (7–12). Three case series (8–10) list neurologic symptoms as the presenting symptoms—although that does not mean sole or isolated CNS disease—in 4% of cases each. The overall incidence and the proportion of truly isolated CNS Whipple’s disease are too rare and too small to allow for any meaningful estimates. Patients who are symptomatic may be only a small subset of those with bacterial infection in the CNS or with CNS pathology. This follows from two types of observations. First, a PCR-based study (69) demonstrated positive results for T. whipplei in CSF in 7 out of 10 (70%) neurologically asymptomatic patients at the time of diagnosis of intestinal Whipple’s disease. Second, autopsy cases from the preantibiotic era had a high proportion of pathologic findings in the brain. Sieracki et al. (2) described extensive CNS involvement at autopsy in two patients with Whipple’s disease who never reported CNS symptoms. In a series of postmortem examinations on patients with Whipple’s disease described by Enzinger and Helwig (3), 10 of 11 patients had characteristic brain lesions associated with this disease. It has been suggested that isolated CNS manifestations as primary presentations of Whipple’s disease may be increasing in frequency and that this may be attributed to the incidental use of antibiotics that suppress early, presymptomatic non-CNS disease but do not penetrate well enough into the CNS to halt progression there (70). Again, the numbers of isolated CNS cases are insufficient for a careful analysis of this issue, and increasing disease awareness may be an alternative explanation.
PATHOGENESIS AND PATHOPHYSIOLOGY
The pathogenesis of Whipple’s disease and of T. whipplei infection is an evolving field. A number of immunologic abnormalities have been identified in patients with Whipple’s disease, and these are presumed to play a role in pathogenesis by way of increasing host susceptibility to infection. However, so far these abnormalities are all of a quantitative nature: No clear genetic defects or dichotomous presence or absence of any defined phenotypic markers have been described. Identified abnormalities include (a) diminished ability of macrophages of Whipple’s disease patients to degrade intracellular microorganisms (71–73); (b) reduced interleukin-12 production by patients’ peripheral blood monocytes upon stimulation with bacterial antigens (74); (c) dysregulation of mononuclear cell function such that the components of a T helper 1 (Th1) immune response are reduced and those of a T helper 2 (Th2) immune response increased (75); (d) diminished Th1 reactivity of peripheral and duodenal CD4+ T cells upon stimulation with specific T. whipplei antigens (76); (e) a transcriptional pattern of M2/alternatively activated macrophages—which is associated with a Th2 immune response—as detected in duodenal tissue of one patient with Whipple’s disease (77); (f) a lack of an inflammatory response and a predominance of an M2/alternative activation phenotype in peripheral and duodenal macrophages, in combination with reduced nitrite production in duodenal tissue and an impaired capacity of peripheral monocytes to perform oxidative burst upon exposure to T. whipplei (78); and (g) a “paradoxically” higher immunoglobulin G (IgG) antibody response against T. whipplei in asymptomatic carriers as compared to patients, indicating a diminished serologic response to infection (79,80).
There are a few reports of secondary or opportunistic infections in patients with Whipple’s disease (81), but apart from Giardia duodenalis infection, which has been reported in 8% and 12% of patients, respectively, in two case series (82,83), these are not commonly observed phenomena, and humoral immune responses against other pathogens are largely intact (4). Also, there is no significantly increased incidence of Whipple’s disease in patients with known primary immunodeficiencies or immunosuppressive therapies and instead only reports that immunosuppressive therapy may hasten the onset of clinically apparent Whipple’s disease in patients with prodromal articular manifestations (84,85). Surprisingly, one sequence-based study of the lung microbiome (i.e., the lung’s microbial content and composition) (86) found T. whipplei DNA in HIV-infected individuals. Furthermore, in 11 of the 82 HIV-positive subjects, T. whipplei sequences dominated the microbial community (>50% of sequence reads). The clinical significance of these findings is unclear because none of the individuals suffered from respiratory complaints or known lung pathology, but these findings also suggest that T cell–based immunity is important in controlling the organism.
Based on the detection of T. whipplei in acute self-limited illnesses and also in asymptomatic people, it has been proposed that T. whipplei may be transmitted from infected hosts or the environment, possibly in early childhood; may cause acute primary infections in some; may go on to be carried by some; and may cause classical Whipple’s disease only in those who lack the immune response to contain the organism (87,88). However, at this point, available data are not sufficient to be able to confirm these proposals. Taken together, there is good evidence that immunologic factors play an important role in pathogenesis, but the abnormalities identified so far are comparatively modest and mainly manifest as immunologic dysregulation between a Th1 and Th2 response.
Current concepts hold that the disease process is initiated by translocation of the bacterium from the intestinal lumen into the intestinal mucosa, possibly via invasion of intestinal epithelial cells or through epithelial intercellular junctions, followed by crossing of the basement membrane and replication in the intestinal lamina propria (24,89). There, the bacteria are ingested by macrophages and appear to spread via blood vessels or via lymphatics into mesenteric lymph nodes and into the systemic circulation. Infected blood monocytes are possible vehicles for dissemination to other organs (90). Uncertainty still exists concerning the preferred compartment for bacterial multiplication. Earlier electron microscopic studies indicated that most intact bacterial structures in untreated intestinal Whipple’s disease are extracellular, just beneath the intestinal basement membrane, whereas ingested bacteria in macrophages exist both with intact shapes and in varying stages of degradation (24,25). In situ hybridization localized the 16S rRNA of T. whipplei (corresponding to intact bacteria) to the extracellular spaces of the intestinal lamina propria, most concentrated underneath the basement membrane, but not in macrophages (28). However, some investigators also report intracellular replication in a monocyte and macrophage cell culture model and enhanced replication after addition of interleukin-16 (91).
Once T. whipplei disseminates, the brain is one of the favored sites for metastatic infection. The mechanisms by which the bacteria cross the blood–brain barrier and infect brain parenchyma are unknown. Entry by extracellular bacteria or by bacteria within monocytes both are possibilities. The perivascular and subependymal distribution of macrophage nodules suggests bloodborne bacterial dissemination. The distribution of microorganisms and histologic lesions within the CNS is relatively widespread and therefore argues against highly localized microbial tropism. The size and multifocal nature of the microglial nodules in CNS Whipple’s disease is compatible with neurologic deficits described in patients. For example, microscopic lesions in the oculomasticatory segments of the midbrain and upper pons might explain the unusual features of oculomasticatory myorhythmia (OMM) or oculofacial-skeletal myorhythmia (OFSM), both findings that are nearly pathognomonic for CNS Whipple’s disease (92–94). However, the degrees to which CNS manifestations are directly due to the bacteria or to immunologic or cellular responses are not known. In cases with extensive CNS disease, bacteria are found within neurons and in macrophages (see the next section), and neuronal loss and reactive astrocytosis are seen during later stages of the disease process.
PATHOLOGY
In 1936, Ford and Walsh (95) described the case of a 47-year-old man who died after a 10-month history of sleep disturbance, progressive paralysis of ocular movements, keratitis, bulbar palsies, and what would later be called OMM (92). They did not recognize this as a case of Whipple’s disease. Notable findings at postmortem examination included cerebral atrophy, swelling of basal ganglia, and widespread degenerative changes of the cerebral gray matter with encephalitis, especially involving the corticobulbar tracts. In some areas, there was neuronal cell death; other neurons were swollen with a granular bluish purple cytoplasm. More than 50 years later, these tissues were reexamined with a variety of histologic techniques, including the PAS stain (96). The encephalitis involved extensive mononuclear cell infiltration of gray and white matter with perivascular cuffing, microglial proliferation, and marked astrocytosis. Foamy macrophages formed nodules and contained PAS-positive accumulations. Some neurons and astrocytes were PAS positive as well. These are all features that are typical of Whipple’s disease pathology in the brain. In addition, electron microscopy confirmed the presence of bacillary structures in the cerebral cortex as well as in retinal tissue.
Other cases of CNS involvement in Whipple’s disease have probably gone unrecognized (2,3). For example, 3 of the 34 patients with Whipple’s disease that were reported in the literature between 1907 and 1948 had symptoms referable to the CNS at the time of death, including confusion and somnolence (97); however, CNS Whipple’s disease was not formally discussed before 1960. The development of PAS reagents and electron microscopy greatly facilitated this discussion. Sieracki et al. (2) were the first to address CNS pathology in Whipple’s disease directly. Two postmortem examinations revealed numerous and widespread periventricular, subependymal, and subcortical nodules, containing PAS-positive sickle-form particle-containing (SPC) cells. These SPC cells corresponded to the same foamy macrophages that had been traditionally associated with Whipple’s disease in intestinal tissues. They noted less consistent involvement of the choroid plexus and leptomeninges. In the autopsy review by Enzinger and Helwig (3) of Whipple’s disease 3 years later, 10 of 11 cases in which CNS tissue was available displayed the characteristic lesions described earlier.
The gross and microscopic pathology of CNS Whipple’s disease has been summarized by a number of investigators (98–101). In many cases, chalky yellowish white 1- to 2-mm nodules are distributed diffusely throughout the cortical and subcortical, cerebral and cerebellar gray matter, and in subependymal locations. They demonstrate a special predilection for temporal, periventricular, and periaqueductal gray matter, hippocampus, hypothalamus, and basal ganglia. Less common sites include the cerebellum and the thalamic and olivary nuclei. They tend to be perivascular. Other common gross pathologic findings in CNS Whipple’s disease include cortical atrophy and ventricular dilation.
On microscopic examination, nodules are aggregates of primarily macrophages (microglia) that stain strongly positive with the PAS reagents (Figs. 28.2 and 28.3). Reactive and hypertrophic astrocytes are common at the periphery of these nodules; lymphocytes and plasma cells are uncommon. Dual staining with PAS reagent and antibodies directed against glial fibrillary-associated protein (GFAP) indicates astrocytes (Fig. 28.3). In cases with more extensive involvement, individual PAS-positive cells are more diffusely distributed and may infiltrate overlying subarachnoid space (100) and white matter (Fig. 28.4). High-power examination of PAS-stained tissue reveals intracellular and extracellular material with variable appearance, from lacy strands to darker specks and larger irregular granules or clumps (Fig. 28.5). Occasionally, a bacilliform particle can be discerned. In areas with more substantial collections of PAS-positive material, there tends to be neuronal loss and demyelination with vacuolization. Smaller (microscopic) microglial nodules have been described that stain less intensely with the PAS reagents (99). Some reports have described microinfarcts that occur most often in the frontal or occipital lobes (100); it is hypothesized that such infarcts may be caused by small emboli released from cardiac valvular vegetations that were found commonly in Whipple’s disease patients in earlier autopsy series (3,4). The phagocytes associated with these infarcts are not PAS positive.
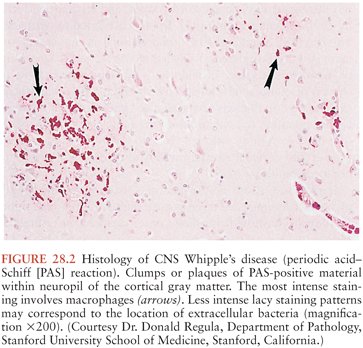
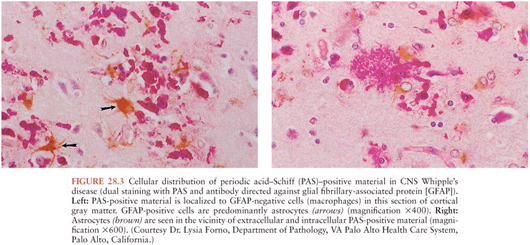
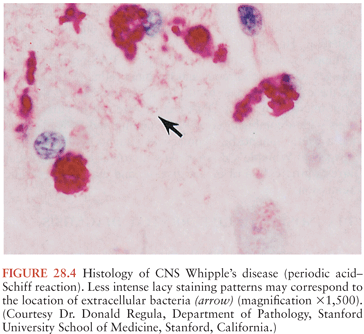
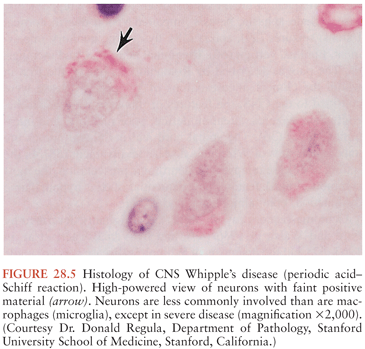
Bacilli were first seen in the brain in 1969 with electron microscopy and resembled those previously described in non-CNS cases of Whipple’s disease (101,102). The distribution of bacteria and bacterial remnants corresponds well with areas of PAS-positive staining. Large numbers of extracellular bacteria can be detected within neuropil and near macrophages. Intact intracellular bacteria, as well as bacilli undergoing various stages of degradation, are found within the widely distributed PAS-positive macrophages (98,101,103–105). Evidence of bacillary binary fission suggests that microbial replication does take place within the CNS (98,101). Macrophages with degraded bacilli found at the center of older lesions stain more strongly with PAS than macrophages with intact bacilli at the periphery (99). Late stages of bacterial degradation produce intracellular accumulations of serpiginous bacterial membranes that create the large, homogeneous, and more strongly staining PAS-positive granules. Whipple’s disease pathology can be focal; involvement of the brain, as with the small intestine, may be patchy (106); thus, a negative random biopsy result does not rule out the disease (see also section “Diagnosis and Differential Diagnosis”).
CLINICAL MANIFESTATIONS
Neurologic manifestations of Whipple’s disease occur in three scenarios: (a) accompanied by intestinal and/or other systemic disease manifestations at the time of diagnosis, particularly when the disease is diagnosed at an advanced stage; (b) in the setting of clinical relapse after antibiotic treatment of intestinal or other manifestations; and (c) as primary or isolated neurologic disease without other apparent manifestations. Among these scenarios, neurologic relapse after treatment (b) is most commonly encountered. This scenario was particularly common before the 1980s, when tetracyclines—which do not cross the blood–brain barrier well—were typically used for treatment. In one review of 88 mostly tetracycline-treated patients, 31 relapsed after initial treatment, and relapses in 13 patients (15% of all patients) affected the CNS (5). CNS relapses generally have a poor prognosis, and some are refractory to renewed antibiotic treatment (4,5,17,107). However, more recent case series (10,12,108,109) show a much lower incidence of CNS relapses, particularly when initial intravenous induction therapy with β-lactams and/or oral long-term treatment with trimethoprim-sulfamethoxazole (TMP-SMX)—which cross the blood–brain barrier better than tetracyclines—have been used.
Presentation with isolated CNS Whipple’s disease is an extremely rare clinical event. However, this may be difficult to differentiate from CNS disease combined with other systemic manifestations; for example, a history of migratory arthralgias or chronic cough might represent extraintestinal Whipple’s disease, but these complaints are quite nonspecific and may not be viewed as significant during the time leading up to more prominent manifestations. Even cases with classic features of Whipple’s disease may be missed because of the drawn-out time course of the illness. Nonetheless, there are a number of notable reports in this context. Some reports represent primary presentation with isolated CNS Whipple’s disease where examination of other organs yielded negative results (65,99,110,111). Several other cases represent primary neurologic presentation where other organs were not investigated (112,113) or where involvement of other organs was also found but was not prominent (92,96,98,114–119). A few published cases represent CNS relapses years after initial treatment, at a time when the non-CNS findings had become completely negative (120–122). Several combined case reports and literature reviews specifically dedicated to primary or isolated CNS Whipple’s disease have been published (63–65); the most recent one by Mohamed et al. (65) discusses 24 cases. However, these published cases should be interpreted with caution, because a significant number of these diagnoses were based solely on PAS-positive histologic findings that have not been confirmed with either electron microscopy or well-validated PCR assays. As discussed (see “Epidemiology” and “Diagnosis and Differential Diagnosis”), PAS-positive histologic findings are not entirely specific for Whipple’s disease. Another report (37) summarizes 20 cases (authors’ own and cases from the literature) that are described as T. whipplei chronic encephalitis, but the case definition used would also fit the description of primary or isolated CNS Whipple’s disease as commonly used by other authors; similar caution concerning the certainty of diagnoses is also indicated with this series.
Common neurologic manifestations of Whipple’s disease are dementia (or cognitive impairment), ophthalmoplegia, myoclonus, altered level of consciousness, psychiatric abnormalities, hypothalamic dysfunction, and ataxia (4,37,67,68,93) (Table 28.1). The first three are considered a characteristic triad that should suggest the diagnosis of CNS Whipple’s disease, although only a minority of patients (approximately 15%) has all three signs together (4,93). Headache is a common but nonspecific complaint. The dementia is slowly progressive and manifest by memory impairment, confusion, personality change, paranoia, emotional lability, and depression. Approximately 71% of patients with CNS Whipple’s disease show signs of cognitive impairment, and nearly half of these patients also demonstrate psychiatric disturbances (93). Patients have sometimes been misdiagnosed with Alzheimer disease. Nearly all cases of ophthalmoplegia are supranuclear, usually in the form of vertical volitional ophthalmoplegia with preserved involuntary extraocular movements in response to head movements. OMM and OFSM constitute a peculiar mixture of synchronized eye movements and myoclonus that are reported in about 20% of patients; both may be pathognomonic for CNS Whipple’s disease (see later discussion) (93), but other forms of myoclonus can occur without eye involvement or in a dyssynchronous fashion. In a particularly severe case of myoclonus, a patient suffered from synchronized 1-Hz jerks of the right face, pharynx, arm, diaphragm, and calf, causing her entire body to shake (114). Segmental myoclonus involving the facial nerve is also described in the setting of Whipple’s disease (123). Hypothalamic signs reported in this disease include polydipsia, polyphagia, insomnia, and hypersomnia (111,120,121,124). In their review of 28 patients, including 12 with CNS manifestations, Fleming and colleagues (8) found headache, diplopia, depression, confusion, and other forms of altered personality to be common at the time of presentation. A subsequent review of CNS Whipple’s disease cases from the same institution (67) found cerebellar ataxia (6 of 11 cases, 55%) to be more common than described in other series.

Related posts:
 Neurologic Manifestations of Varicella and Herpes Zoster
Neurologic Manifestations of Varicella and Herpes Zoster
 Meningitis and Encephalitis Caused by Mumps Virus
Meningitis and Encephalitis Caused by Mumps Virus
 Human Immunodeficiency Virus
Human Immunodeficiency Virus
 Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
 Subdural Empyema and Suppurative Intracranial Phlebitis
Subdural Empyema and Suppurative Intracranial Phlebitis
 Rickettsioses, Anaplasmoses, and Q Fever
Rickettsioses, Anaplasmoses, and Q Fever
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


