VIRAL MENINGITIS AND ASEPTIC MENINGITIS SYNDROME
JOSÉ R. ROMERO
Viral meningitis can be characterized as a central nervous system (CNS) viral infection with signs of meningeal irritation (neck stiffness, Kernig and/or Brudzinski signs) and cerebrospinal fluid (CSF) pleocytosis but without neurologic dysfunction due to brain parenchymal involvement (1). It differs from viral encephalitis where evidence of brain parenchymal dysfunction is manifested by an altered state of consciousness, change in personality, or other objective signs of neurologic dysfunction (e.g., seizures, cranial nerve palsies, abnormal reflexes, paralysis, etc.). Although it is common to discuss the two as wholly separate entities, it is important to note that overlap between them (i.e., meningoencephalitis) does occur following infection with many viral agents.
Almost 100 years ago, Wallgren (2) introduced the term “acute aseptic meningitis” to describe a short-lived, self-limited, benign CNS syndrome characterized by the acute onset of the signs of meningeal irritation in which examination of the CSF revealed a mononuclear pleocytosis and the absence of bacteria on direct examination and by culture. In addition, no parameningeal process, acute/chronic systemic infectious disease, or community infectious disease could be identified that could produce the syndrome. With advances in diagnostic methodologies, it became evident that multiple infectious agents (e.g., Lyme disease), inflammatory conditions, drugs, environmental agents, and so forth could cause the syndrome.
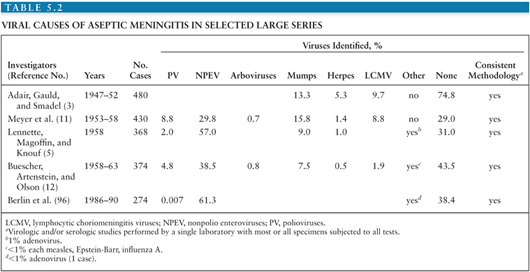
It is estimated that in the United States, the annual number of aseptic meningitis cases is at least 75,000. Viruses account for the overwhelming majority of cases (Table 5.1). Early reports indicated that mumps virus, lymphocytic choriomeningitis virus (LCMV), and poliovirus (PV) were the major identifiable causes of aseptic meningitis (3). As diagnostic techniques improved as a result of the development of cell culture, the enteroviruses were shown to have a major role in causation of syndrome (Table 5.2) (4,5). Nucleic acid amplification tests (NAATs) have bolstered this finding and led to the identification of novel causes (6).
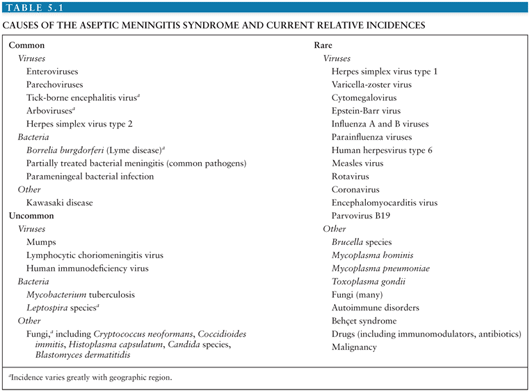
The incidence of aseptic meningitis is influenced by many factors, including effective vaccine programs, sanitation, poverty, and regional endemic viruses (7–10). Previous significant causes of viral meningitis such as PVs, mump virus, and LCMV are now rare or infrequent as a result of effective vaccines, sanitation, or improved housing (3,5,8,11–13). Although many of the infectious causes of aseptic meningitis are reportable (14), the true incidence of the syndrome is unknown due to incomplete reporting, failure to test for specific agents, and because aseptic meningitis is not a reportable condition. In Finland, a 14-year birth cohort study found the annual incidence of viral meningitis in children younger than 14 years of age to be 27.8 per 100,000 (15). A more recent study from Greece documented the annual incidence of aseptic meningitis to be 17 per 100,000 in children younger than 14 years of age (16). Two studies from the United States give widely discrepant estimates of the incidence of aseptic meningitis. A 32-year (1950 to 1981) study from Olmsted County, Minnesota found that the adjusted incidence rate of aseptic meningitis was 10.9 per 100,000 person-years (range 7.9 to 17.8 per 100,000) (8,13). The Centers for Disease Control and Prevention reported that the national incidence for aseptic meningitis ranged from 1.5 to 4.0 per 100,000 for the period spanning 1971 to 1981 (17). The lower incidence in the latter report is most likely the result of passive surveillance and, therefore, underreporting. The incidence of aseptic meningitis is greater in males and in children, particularly those younger than 1 year of age (8,15,16).
ENTEROVIRUSES
Virology and Pathogenesis
The enteroviruses (EVs) are one of six genera (Enterovirus, Cardiovirus, Cosavirus, Hepatovirus, Parechovirus, and Kobuvirus) in the Picornaviridae (pico, “small”; rna, “ribonucleic acid”; viridae, “viruses”) family of viruses known to cause disease in humans. The original classification of the EVs, based on evidence of human origin, pathology in animal models, patterns of replication in cell culture, and physicochemical characteristics, identified 72 serotypes (18). This schema, although initially useful, resulted to the misclassification of several EV and inclusion of several non-EV into the genus (19). The development of experimental and computational methodologies for the study of the molecular biology and genomic analysis of the EV allowed for refinement in the classification of and identification of the EVs.
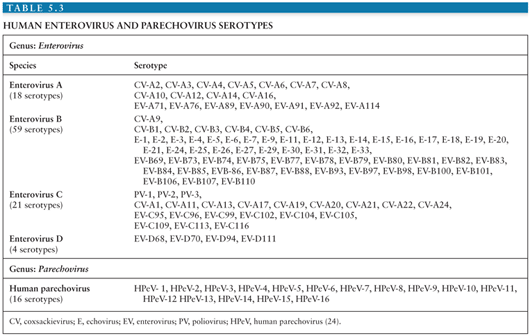
Currently, identification and classification of the EVs is based on the nucleotide sequence of VP1, the largest and most surface exposed of the viral capsid proteins containing important neutralizing epitopes (20–23). Using this approach, the EVs have been speciated into four groups (enterovirus A to D) containing more than 100 serotypes (Table 5.3) (24). In addition, this approach has revealed that several of the “traditional” EV serotypes are actually strains of the same serotype or are not genetically related to the EVs (echoviruses 22 and 23) (20,21,25–27).
The EVs are nonenveloped viruses 30 nm in diameter with a buoyant density of 1.30 to 1.34 g/cm−3 in caesium chloride (CsCl) (19). The lack of an envelope confers to them relative environmental stability where they can survive for days at room temperature. Infectivity can be preserved for weeks at −20°C or with little or no loss of infectivity for years when stored at −70°C. Similarly, the lack of a lipid envelope renders them insusceptible to ether, chloroform, and alcohol. The EVs are inactivated by heating to greater than or equal to 50°C, chlorine, and formaldehyde.
The capsid of all EVs is composed of 60 units each of four structural or capsid proteins: VP1 to VP4, alternatively known as 1A to 1D, arranged so as to give the virion icosahedral symmetry (28–33). Each of proteins VP1 to VP3 is wedge-shaped and composed of an eight-stranded antiparallel β-barrel core. Each of the stands is connected to the next by intervening loops that determine antigenicity, receptor specificity, and confer capsid stability (34). The basic structural element of the viral capsid, the protomer, is initially composed of the proteins VP0, VP1, and VP3 (35). Five protomers self-assemble to form a pentamer. Twelve pentamers, in turn, assemble around a single strand of viral RNA to produce the immature virion. The cleavage of VP0 to yield VP2 and VP4 completes the formation of the mature virion. VP1 to VP3, and in particular VP1, have surface-exposed amino acids which determine the antigenic diversity and the receptor specificity of the EV (20,33,36,37). VP4 is not surface exposed but shares close association with the viral RNA and plays a vital role in release of the genome after viral attachment (38).
The surface topographies of the various EVs share a number of similarities. These include a plateau or mesa located at the fivefold axis of symmetry formed by the union of five protomers. Surrounding this plateau is a deep cleft or canyon into which a viral receptor inserts when the EV encounters a susceptible host cell (36). Additionally, the host immune response to EV infection generates serotype-specific antibodies directed to antigenic sites around the fivefold axis and canyon walls, thus blocking viral-host receptor interaction and infection. Lastly, beneath the canyon floor exists a hydrophobic pocket containing a lipophilic factor. This pocket has been the target for the development of anti-EV drugs that result in altered receptor binding and viral uncoating (39,40).
The EV genome consists of single-stranded, positive (messenger)-sense RNA of approximately 7,400 nucleotides (nts) in length. The genome layout may be summarized as follows: VPg+5′UTR[1A-1B-1C-1D/2A-2B-2C/3A-3B-3C-3D] 3′UTR-poly(A) (24,41). The 5′ end of the genome is covalently linked to a small protein, VPg, essential for viral RNA replication. The genome is organized into a long 5′ untranslated region (5′ UTR) of approximately 740 nts that immediately precedes a single open reading frame (ORF). The ORF measures approximately 6,630 nts and is followed by a short (approximately 70 nts) 3′ UTR and a terminal polyadenylated tail.
The 5′ UTR contains multiple regions of predicted higher order structure and highly conserved nucleotide identity among the EV. This region of the genome contains elements essential for viral RNA replication, translation of ORF, and, in the PVs, determinants of neurovirulence. Because of the highly conserved nucleotide sequences within the 5′ UTR found among all the EV, it serves as the target for NAATs for the detection of the EV now in common use (42,43).
Translation of the ORF by host cell ribosomes is accomplished in a nonconical, cap-independent manner giving rise to a single polyprotein that is posttranslationally cleaved by viral and host proteinases to yield 11 viral proteins (four structural and seven nonstructural) as well as several functional protein intermediates. The ORF can be subdivided into three regions: P1 to P3. The P1 region encodes for the four structural proteins (VP1 to VP4 or 1A to 1D) that comprise the viral capsid. These are organized 5′ to 3′ as VP4 (1A), VP2 (1B), VP3 (1C), and VP4 (1D). The P2 and P3 regions code for seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) that are essential for the viral life cycle. The intermediate proteins play roles in viral replication.
Immediately downstream of the ORF is a short 3′ UTR followed by a terminal poly(A) tract. Similar to the 5′ UTR, the 3′ UTR is predicted to have higher order structures and play a role in genome replication (44).
Following binding of the EV to a host cell receptor, conformational changes in the virion result in release of the viral genome into the cytoplasm of the host cell. The RNA genome is replicated through a double-stranded RNA intermediate that is formed by the EV RNA-dependent RNA polymerase (3D).
Despite more than 100 years of study of the pathogenesis of EV infections, much still needs to be learned. Most of what is known stems for the study of PV types 1 to 3 using information derived from human disease and nonhuman primate, murine, and, most recently, transgenic (Tg) mouse models expressing the PV receptor, PVR or CD155 (45–50). It is likely that the non-PV EVs share similar mechanisms of pathogenesis.
The majority of the EVs are transmitted via a fecal-oral route. In addition to direct person-to-person or fecal-oral transmission, experimental or clinical evidence exists for the transmission of the EV via houseflies, housefly-contaminated food, water, and sewage (51–54). Bivalves have been found to accumulate EV (55,56). However, their role in transmission has not been established. Notable exceptions to fecal-oral transmission include coxsackievirus (CV)-A21, EV-D68, and EV-D70, which may spread via contaminated fomites or ocular and respiratory secretions. Evidence for transplacental transmission exists, leading to congenital infection (57–61).
Following ingestion of the EVs, infection of the cells of the nasopharynx and, more significantly, the lower gastrointestinal (GI) tract occurs. The inherent acid-resistance of the EV favors the latter. Replication in the lymphatic tissues of these sites (i.e., tonsils and Peyer patches) leads to shedding of the EV from the nasopharynx for 1 to 2 weeks and from the GI tract in feces for several weeks to months (7). Seeding of EV to the deep cervical and mesenteric lymph nodes ensues and results in their spread to the systemic circulation via the lymphatics. This primary or minor viremia leads to seeding of various organs, including the CNS, liver, lungs, skin, and heart. Further replication in the tissues of those organs results in a major (secondary) viremia. If the CNS was not seeded during the initial viremic episode, spread there may occur with the major viremia. Viremia or presence of virus in the CNS continues until the host develops type-specific neutralizing antibodies directed to the capsid proteins, usually by day 7 to 10 postinfection. Immunoglobulin A (IgA) antibodies appear in the respiratory and GI tracts 2 to 4 weeks after infection. Unlike other viruses, which are largely contained by cellular immune mechanisms, the EVs are cleared from the host primarily by antibody-mediated mechanisms.
Great strides have been made in understanding the pathogenesis of EV infections at a molecular level. The presence of an EV receptor is the primary, but not the sole, determinant for cellular infection (62). As stated previously, PVR has been shown to be the receptor for the PVs (62) and maps to chromosome 19. PVR is a member of the immunoglobin superfamily and functions as an adhesion molecule. It helps to form adherens junctions and is a recognition molecule for natural killer cells. In addition to PVR, at least 11 other cell proteins have been identified as receptors or coreceptors for other EV serotypes (Table 5.2) (63–66).
The exact identity of the cells in the upper and lower intestinal tract infected by the PVs is unknown. However, they have been identified within the ileal wall and mesenteric nodes in human infection (45,49,50). It is believed that the PV infect either lower GI cells expressing the PVR or use transcytosis through microfold (M) cells in the lower GI tract to gain access to lymphoid tissue. Supporting this is the finding of PVR on the surface of intestinal epithelium, M cells, and in the germinal centers of Peyer patches (67). Support for the latter comes from the finding that M cells can bind and endocytose PVs (68,69).
Why the majority of EV infections do not result in clinically apparent infection (70–73) comes for a number of studies that suggest that replication of PV in extraneural tissues is inhibited by the host interferon (IFN) response. PVR Tg mice that are IFN α/β receptor deficient are highly susceptible to PV infection, and PV replication in the small intestine is enhanced (74,75). Thus, IFN responses may be crucial in limiting the spread of EV infection.
Lastly, the mechanism of EV entry into the CNS remains unclear. Evidence for two pathways exist for PV: transit through the blood–brain barrier (BBB) or via retrograde axonal transport. For the development of paralytic disease in chimpanzees, viremia has been shown to be essential (76). This finding provided initial support that the BBB may be a route to the CNS. Endothelial cells may express EV receptors that may influence tissue susceptibility to the EV (77) and facilitate virus entry into the CNS and other organs. Further buttressing of this hypothesis came from the finding that cultured human brain microvascular cells can support PV replication (78,79). However, in other studies, pharmacokinetic analysis of PV injected into Tg and non-Tg mice indicated that PV was delivered to the brain in significantly greater amounts than would be expected from the vascular concentration (80). This suggests that PV may enter the CNS via the BBB but without need of PVR.
Evidence in humans supports EV access to the CNS via a neural route. Individuals inadvertently inoculated with an incompletely inactivated PV vaccine developed initial paralysis in the limb receiving the vaccine (81). Trauma to a limb preceding PV infection has been associated with the development of paralysis of that limb. “Provocation poliomyelitis” following intramuscular injections into an extremity of a person incubating wild type PV or those receiving live attenuated PV vaccines has been well documented (82,83).
Substantial evidence from nonhuman primate and murine models exists in support of EV access to the CNS via a neural route. In monkeys, inoculation of the sciatic nerve with PV results in viral spread along the inoculated nerve and the spinal cord (84). The initial limb to develop paralysis following intramuscular inoculation of PV in monkeys and Tg mice is the one injected (85–87). If the sciatic nerve of the intramuscularly inoculated lower extremity is frozen or transected, paralysis of the limb is prevented (84,87). In Tg mice, it has been shown that in provocation PV, there appears to be induction of retrograde axonal transport (88). PV may gain access to neurons at the level of the neuromuscular junction.
The search for viral genomic determinants of neurotropism and neurovirulence has focused on the PVs (89). After immunization with live, neuroattenuated vaccine (Sabin) PV strains, shedding of PV that has recovered the ability to cause paralysis (i.e., neurovirulent revertant strains) occurs routinely. Comparison of wild type, vaccine, and revertant PV strains has identified a 10-nucleotide region within the 5′ UTR (nts 472 to 484 relative to PV type 3 Sabin strain) where neuroattenuating mutations are found to cluster. Additional minor determinants of virulence are localized to amino acids encoded in the P1 and P3 regions. The search for determinants of neurovirulence in other EV has been unsuccessful.
Histopathology
The benign nature of EV meningitis has made human pathologic data for this disease sparse. A report of a child who died of CV-B5 myocarditis with concomitant meningitis describes inflammation of the choroid plexus of the lateral and fourth ventricles, fibrosis of the vascular walls with focal destruction of the ependymal lining, and fibrotic basal leptomeninges (90). Parenchymal findings were limited to moderate symmetric dilation of the ventricles and an increase in the number and size of subependymal astrocytes. The inflammatory reaction at the choroid plexus supports the concept of viremic spread to the CNS. An adolescent presenting with a similar constellation of findings died of systemic CV-B3 infection (91). The dura was grossly distended with swelling also of the pia, arachnoid, and brain parenchyma. Microscopically, round cell infiltrates were noted in the meninges overlying the cerebellum; the brain parenchyma was congested with increased numbers of oligodendrocytes. Lymphocytic infiltration was most prominent around blood vessels in the cerebral white matter and in the basal ganglia, again suggesting viremic CNS access; focal areas of necrosis and hemorrhage were also seen (91).
Epidemiology
The EVs have a ubiquitous worldwide distribution; humans are their only natural reservoir (58,92). It is estimated that they result in more than 1 billion annual infections worldwide (22). In the United States alone, the non-PV EVs are estimated to cause 30 to 50 million infections each year. Due to underreporting of EV infections (93), a well-grounded estimate of the number of cases of EV meningitis that occurs each year is not possible. However, conservative estimates place the annual number to be between 30,0000 and 75,000 cases (22,94). The EVs are responsible for 80% to 90% of identifiable causes of viral meningitis (94–97). Wild type PV were eradicated for the West Hemisphere in 1991 (98) and are currently endemic only in Afghanistan, Nigeria, and Pakistan (99). As such, they do not contribute to the burden to EV in most of the world.
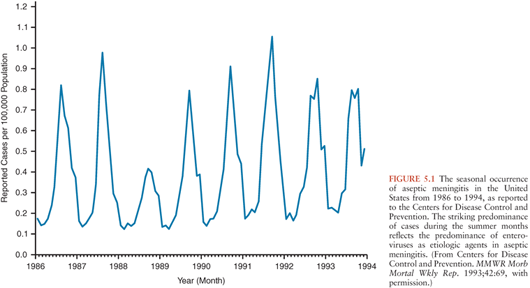
In regions with temperate climates, the non-PV EV exhibit marked seasonality with the majority of infections occurring in the summer and fall (Fig. 5.1) (92,100,101). This being said, EV infections do occur during the winter, warranting their inclusion in the diagnostic evaluation of aseptic meningitis during that time of year (102–106). In tropical and subtropical areas, EVs occur year-round, but with higher incidence during the rainy season.
Despite the existence of more than 100 serotypes of EV, only a limited number are responsible for the majority of disease observed annually in each geographic region (58,93,107–110). The rank of each serotype within the most frequently isolated EVs varies annually and geographically. In the United States, 15 serotypes accounted for approximately 80% of all EVs reported from 1970 to 2005 (in descending order): echoviruses (E)-9, -11, -30, CV-B5, E-6, CV-B2, CV-A9, E-4, CV-B3, E-7, CV-B4, E-18, CV-B1, E-3, and -5 (93). Some serotypes cycle with varying periodicity within a community (58,93,109,110), a reflection of the availability of new susceptible host populations (especially children). Other serotypes appear de novo as novel epidemic-associated viruses (111). Around the world, the serotypes most commonly isolated from the CSF and, therefore, from cases of meningitis or meningoencephalitis, belong to the Enterovirus B species (93,108,112,93,113,–114). In the United States, the serotypes most frequently isolated from CSF specimens over a 36-year period, in descending order of frequency, are E-9, -11, -30, CV-B5, E-6, CV-B2, CV-A9, E-4, CV-B4, E-7, -18, and -5 (93).
Outbreaks of EV meningitis are common. Large nation- and community-wide outbreaks involving thousands of individuals have been well documented (115,116). Outbreaks involving more localized venues such as neonatal units, nurseries, daycare centers, orphanages, schools, pools, camps, and sports teams occur (53,117–122). Sequential episodes of EV meningitis involving different serotypes have been reported to occur within a month of each other (123–125). Mixed infections involving EVs, other viruses, or bacteria have been well described (102–130).
Children represent the overwhelming majority of cases of EV meningitis. An incidence peak among young infants and school-aged children ages 5 to 10 years has been reported in multiple studies (58,131–133). Occasional outbreaks of EV CNS infections occur predominantly among adults (134–136). A possible explanation for these findings may lie in the history of the particular serotypes in the geographic area studied. Serotypes with “endemic” patterns, those occurring with significant incidence annually, are most likely to affect only the youngest children because of their absence of previous exposure and immunity. Older children and adults are more likely to predominate in an outbreak of a serotype that has not been present in a community for several years, thereby creating a reservoir of susceptible individuals among children born since the last appearance of that serotype.
Host factors that predispose to EV meningitis, other than young age and immunodeficiency, have been difficult to identify. A slight male-to-female predominance in the incidence ratio for EV infections has been noted in large series. However, in a recent report, male predominance was present only among persons younger than 20 years of age (male/female ratio 1.4:0.9) (101). This most likely represents the larger number of females exposed to young children who are principal source of household exposure. Infection rates are higher among persons of lower socioeconomic status, in areas of crowding, and larger families (7).
Clinical Manifestations
The clinical manifestations of aseptic meningitis do not significantly differ among the non-PV EVs causing the syndrome (9,137). However, clinical manifestations do vary with the age and immune status of the patient. Meningitis or meningoencephalitis, singularly or in combination with other syndromes, are common manifestations of symptomatic EV infection in neonates and young infants. Two large retrospective reviews have documented that 62% of infants younger than 3 months of age with group B coxsackievirus infections and 27% of neonates younger than 2 weeks of age with infections due to the echoviruses had associated meningitis or meningoencephalitis (60,138). In two prospective studies, clinical or laboratory evidence of meningitis was found in 42% to 75% of neonates with EV infection (139,140).
Early presentation of EV infection following birth suggests transplacental, intrapartum, or immediate postpartum acquisition of virus (60,138,139). Maternal illness (fever, symptoms of upper respiratory tract infection, abdominal pain) has been reported to occur in 14% to 68% of infected neonates (60,138–141). In neonates, fever (≥38.0°C) is almost universal and accompanied by any or all of the following nonspecific signs: irritability, lethargy, poor feeding, emesis, and upper respiratory tract findings (60,139,140,142). On physical examination, the fontanelle may be full or bulging. Signs of meningeal irritation such as nuchal rigidity, Brudzinski, and Kernig signs are generally absent. An exanthem may be present. If encephalitis in addition to meningitis (meningoencephalitis) is present, the neonate may present with profound lethargy, seizures, and focal neurologic abnormalities that suggest herpes simplex virus (HSV) infection. In some newborns, encephalohepatomyocarditis syndrome may develop in which signs and symptoms of severe hepatitis and myocarditis are superimposed on those of meningoencephalitis (143). Disseminated intravascular coagulation and other findings of “sepsis” result in an illness that may be indistinguishable from that caused by overwhelming bacterial infection.
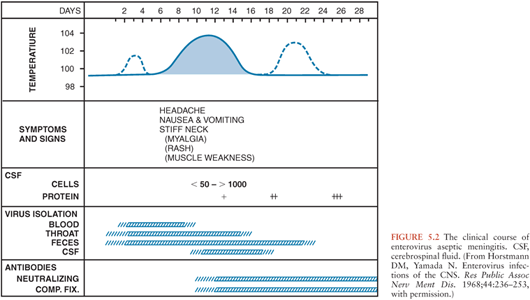
In infants and children, following an incubation period of 5 to 10 days, the onset of EV meningitis is usually abrupt with fever (38° to 40°C), the most common presenting sign (137,143–147). The natural history of EV meningitis is depicted in Figure 5.2. The fever pattern may be biphasic, initially appearing in association with nonspecific constitutional signs and symptoms followed by resolution and subsequently reappearing with the onset of meningeal signs (137,148). Headache is nearly always present in those individuals old enough to report it. Interestingly, it may be ameliorated following the performance of a lumbar puncture, indicating that it may be the result of increased intracranial pressure (149,150). Photophobia is commonly reported. Infants and young children may be irritable or, less commonly, lethargic (142). Nonspecific findings, singly or in combinations, include anorexia, exanthems, malaise, sore throat, abdominal pain, nausea, emesis, and myalgias (144,147). In infants, the fontanelle may be full or bulging. Less than 5% of infants younger than 3 months of age have signs of meningeal irritation (142). However, these become more common in older patients (144,147,151,152). Seizures are noted in less than 5% of children (142,146). Other uncommon complications include coma, increased intracranial pressure, and inappropriate secretion of antidiuretic hormone (142,153). The duration of EV meningitis in infants and children is generally less than 1 week.
Fewer clinical reports exist documenting the presentation of EV meningitis in adolescents and adults (134,136,154–157). Headache is the most frequently reported symptom and is nearly uniformly present. The severity of the headache may be such as warrant the use of narcotic analgesics in order to control the pain (157). Photophobia, fever, signs of meningeal irritation, nausea, emesis, and neck stiffness occur in more than 60% of cases. Other less frequently encountered signs and symptoms include myalgia, exanthems, and abdominal pain. Full recovery takes longer in adolescents and adults and may require up to 2.5 weeks (157).
In individuals with humoral immunodeficiencies (X-linked agammaglobulinemia, X-linked hyper IgM syndrome, common variable immunodeficiency), EV infection may result in chronic meningitis or meningoencephalitis that may last for years and often have a fatal outcome (158–161). In addition to the common signs and symptoms of EV meningitis mentioned previously, neurologic manifestations include paralysis/paresis, seizures, cognitive impairment, developmental regression, sensorineural hearing loss, coma, dysarthria, hydrocephalus, and aphasia. Extra CNS manifestations occur singly or in combination in a significant number of cases and include dermatomyositis, chronic hepatitis, arthritis, myocarditis, and subacute lumbosacral polyradiculopathy. In patients who have undergone repetitive, sequential lumbar punctures, the cell culture detection of EV in CSF has been intermittent. However, using NAATs, evidence of their persistence in CSF has been documented (162). Treatment with antibody preparations intravenously and intrathecally or intraventricularly has resulted in stabilization of some of these patients; however, viral persistence has been documented during therapy (159,162,163). With the availability of intravenous preparations of immune globulin and the early recognition of this illness, fewer patients appear to be progressing to the classic description of this disease, and atypical neurologic presentations have appeared. The mortality rate in patients with humoral immunodeficiencies may be as high as 50%.
The infected neonate is at greatest risk of severe morbidity and mortality when signs and symptoms develop in the first days of life (60,138,139,164). Neonates infected with CV-B4 were found to be higher risk of death than those infected with other EV. The short-term prognosis of young children with EV meningitis early in life appears to be good; however, there has been some controversy over possible later sequelae. Neurologic, cognitive, developmental, and language abnormalities have been reported in controlled studies of long-term outcome in children with EV meningitis during infancy (165–168). In the largest and most meticulously controlled study, however, no differences between patients and controls could be demonstrated in any of the neurodevelopmental parameters studied (142). Less well studied are the ultimate outcomes of aseptic meningitis cases in older children and adolescents; preliminary data suggest possible school and learning difficulties, but control patients were not studied (169).
Laboratory Findings and Diagnosis
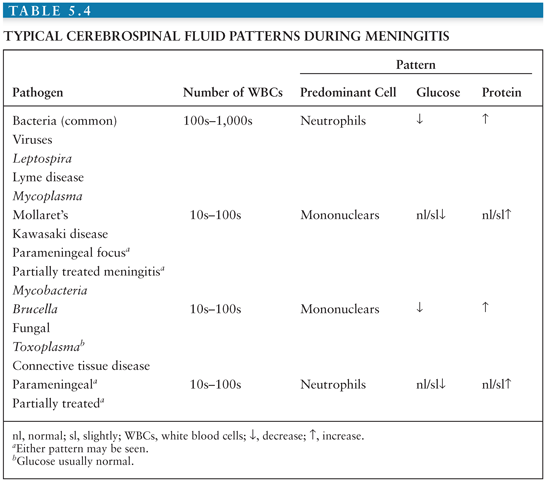
Salient among the laboratory analyses performed for the evaluation of EV meningitis is the evaluation of the CSF. CSF analysis can provide initial clues as to the etiology of the clinical syndrome (Table 5.4). Cytochemical analysis of the CSF typically shows a mild to moderate lymphocytic pleocytosis ranging from 10 to 1,000 cells/mm3 (136,139,141,142,145,149,160). White blood cell (WBC) counts exceeding 1,000 cells/mm3 are seen occasionally (60,103,147,170). Although pleocytosis is almost always present, EVs have been isolated by cell culture or detected by NAAT from the CSF of patients with clinical evidence of meningitis without pleocytosis (171). This is particularly true in the young infant. If the CSF is examined early in the course of the illness, a predominantly polymorphonuclear pleocytosis may be observed (136,139, 142,146,149). Reexamination of the CSF several hours later will document a typical lymphocytic pleocytosis (172–174). The progression of an initially polymorphonuclear pleocytosis to one of a more viral meningitis mononuclear pleocytosis has also been observed with St. Louis encephalitis virus (175). The CSF protein concentration is mildly to moderately increased, whereas the glucose concentration is generally normal. However, hypoglycorrhachia may occur, serving to confound the assessment by suggesting a bacterial etiology (146,147,170).
Traditionally, the diagnosis of EV meningitis has relied on isolation of the virus from CSF using cell culture or inoculation of suckling mice (176). Although initially very useful, these techniques have significant limitations. The sensitivity of tissue culture for EVs is only 65% to 75% (177). The titer of EVs in the CSF of patients with aseptic meningitis may be as low as 101 to 103 TCID50 (median tissue culture infectious dose) per milliliter of CSF. This results in slower growth than is observed with specimens of throat or rectal origin. The time to isolation of EV from CSF ranges from 4 to 8 days (178) using traditional cell culture—too long to be clinically useful in patient management. Using shell vial culture, the time can be shortened to 2 to 3 days, but sensitivity may be lost (179,180). Lastly, optimum recovery of EVs from clinical specimens requires the use of multiple cell lines, either individually or as mixtures, in order to increase culture yield (181). Even using multiple cell lines, some EVs, in particular the group A CVs and some of the newer EVs, fail to grow in cell culture (182). The added cost and technical expertise required for suckling mouse inoculation makes it impractical for use in the modern diagnostic laboratory. Serologic confirmation of EV infection is also generally impractical and not useful in acute management of the patient.
As mentioned previously, the 5′ UTR contains regions of conserved nucleotide identity among the EVs. These regions have been exploited for the creation of primers and probes that can be used in NAATs capable of detecting all EVs (43). Compared to cell culture, NAAT detection of EVs in the CSF has been shown to exhibit sensitivities that range from 86% to 100% and specificities ranging from 92% to 100%. Furthermore, NAATs are capable of detecting EV genome in CSF samples from individuals with syndromes clinically compatible with aseptic meningitis previously deemed negative by cell culture or without pleocytosis (163,162,183). These assays are also able to detect EV that cannot be grown in cell culture. Lastly, NAAT can be performed rapidly, generally in a matter of hours. The results can be made available with sufficient speed as to have an impact on patient management, resulting in a reduction in hospital stay, antibiotic use, and ordering of ancillary tests (184–188). A confirmatory polymerase chain reaction (PCR) test result obtained on a patient with clinical aseptic meningitis can reassure the clinician that no further diagnostic investigation is required. For these reasons, NAAT detection has become the method of choice for the diagnosis of CNS EV infection. Because of the lack of sensitivity of viral culture for detection of the EV in CSF, it should be reserved for instances when NAAT is not available.
Two caveats should be borne in mind when establishing the diagnosis of EV meningitis. Confirmation of EV as the etiology for aseptic meningitis syndrome should rely on detection of the virus from the CSF. As previously discussed, following EV infection viral shedding may occur from the throat and GI tract for up to several weeks (7,92). Therefore, detection of the EV from the stool or throat of an individual with aseptic meningitis may represent an infection that occurred weeks previously and is unrelated to the present syndrome. Shedding from a past infection cannot be ruled out unless the virus is detected in nonpermissive sites (i.e., CSF, blood, tissue) (189).
Lastly, rare reports of co-infections of the CSF by bacteria and EVs exist (102,126,129,130). In these patients, the clinical and laboratory picture of bacterial meningitis dominated and the virus was isolated incidentally. The patients were sick enough that identification of a virus before identification of the bacterium would have been unlikely to dissuade the clinician from continued use of antibiotics. Thus, the detection of an EV either by culture or NAAT must always be placed in the context of the patient’s clinical picture and laboratory findings. In the clinical presentation typical of viral meningitis, co-infection with a clinically “silent” bacterium would be extraordinarily unlikely.
Treatment and Prevention
No specific treatment exists for EV meningitis. Supportive measures include bed rest, antipyretics, and analgesics, as indicated. Administration of parenteral fluids for individuals unable to take adequate fluids orally, especially infants, is indicated. Seizures should be controlled with appropriate anticonvulsant drugs.
Immune globulin preparations have been used for the treatment of newborn infants with severe disease and immunocompromised individuals, but their efficacy is not established (61,159,190). Intravenous, as well as intrathecal, administration may be necessary to ameliorate or prevent CNS infection in immunocompromised patients.
The promising results from clinical trials of pleconaril (39), an antiviral that inhibits EV binding and viral uncoating, were overshadowed by its adverse effects (40). In clinical trials, pleconaril was found to induce cytochrome P450 3A, resulting in menstrual irregularities in women taking hormonal contraceptives. This finding raised concerns that it might increase the metabolism of some hormonal contraceptives and anti-HIV drugs, thereby reducing their efficacy, prompting the U.S. Food and Drug Administration not to grant a license for its use.
The EVs are spread primarily through a lack of good hygiene. Hand washing prevents the spread of the EVs and should be encouraged in families and institutions (191). In patients hospitalized with EV, meningitis infection control measures using standard precautions are sufficient. Community measures for the prevention of EV infections rely on the development and maintenance of sewage and potable water systems. No vaccines exist for the non-PV EV. However, recent early studies suggest that it may be possible to develop an inactivated EV-A71 vaccine that can induce neutralizing antibodies and is well tolerated in humans (192).
PARECHOVIRUSES
The first two members of the genus human parechoviruses (HPeVs) were found in 1956 (193). However, it was not until the turn of the century that they were accorded their own genus. Originally classified as EVs and designated as echoviruses 22 and 23 (18) (currently designated HPeV 1 and 2, respectively), it became evident early on that they exhibited characteristics that differed from the other members of the genus (193,194). The development of methodologies to probe the molecular aspects of viral replication and viral genetics confirmed that they differed substantively from the EV (25–27,195,196) and led to their reclassification in a separate genus (24).
Morphologically, HPeVs exhibit similar characteristics as the EV: small size, lack of an envelope, a positive sense, and single-stranded RNA genome of the length and organization consistent with that of the EVs (6). Important differences are the lack of maturational cleavage of VP0 to yield capsid proteins VP2 and VP4. As a result, the HPeV capsid is composed of three, rather than four, proteins. Differences also exist in the function of two nonstructural proteins. A detailed discussion of these and other differences is beyond the scope of this chapter, and the reader is directed to a recent review (6).
The genus is currently composed of 16 serotypes (Table 5.3) (24). HPeVs have been reported worldwide. The epidemiology of the HPeV continues to evolve as new serotypes are identified and detection is improved using NAATs. Current data indicates that the HPeVs account for approximately 2% of the “EV” isolated using traditional cell culture in clinical laboratories (6). HPeV1 followed by HPeV3 are the types most frequently isolated. Infection with HPeV appears to occur early in life. In the United States, 73% of HPeV1 and 67.6% of HPeV2 isolated come from infants younger than 1 year of age. A longitudinal study of Norwegian infants documented that the cumulative incidence of HPeV infection by 24 and 36 months of age was 86% and 94%, respectively (197).
HPeV infections exhibit a strong seasonal epidemiology. Worldwide, the peak incidence of infections occurs during the summer and fall months (197–204). A unique biennial pattern of circulation has been reported for HPeV3 (205–208). As with the EVs, multiple HPeV serotypes circulate within a community at the same time. The majority of cases of HPeV meningitis occur in male infants younger than 3 months of age. HPeV3 is the overwhelmingly dominant cause of HPeV meningitis. Considerable variation in the annual prevalence of HPeV meningitis is observed.
Transmission of the HPeV occurs via the fecal-oral and respiratory routes. They may be shed from these sites for weeks to months (197,201,209). The finding of HPeV in the stool of healthy, asymptomatic infants indicates that many, if not the majority, of infections are subclinical.
Irritability is present in nearly all cases of HPeV meningitis (202,207,210). An exanthem is frequently present. Emesis, diarrhea, and distention are reported in approximately one-quarter to half of cases. Rhinorrhea, cough, tachypnea, apnea, and wheezing may be present. Notably, findings of increased intracranial pressure (bulging fontanelle) or meningeal irritation (nuchal stiffness, Kernig or Brudzinski signs) are absent. The CSF cytochemical evaluation reveals no or minimal abnormalities in WBC count or protein and glucose concentrations in the overwhelming majority of patients (202,207,210).
Currently, NAAT is the methodology of choice for HPeV detection because of its sensitivity and ability to detect all known HPeV types in a clinically meaningful time frame (6). Optimum diagnostic assays target the HPeV 5′ UTR and are not type-specific. They are designed for increased sensitivity and to broadly detect all HPeV types from clinical specimens.
The HPeV can produce cytopathic effect on appropriate cell lines (211). However, cell culture detection is limited by those factors discussed for the EV (see previous discussion) (211,212).
ARBOVIRUSES
The arboviruses are a group of more than 500 viruses from various viral families that are transmitted by the bite of an insect or tick (i.e., an arthropod vector) (213); hence, the derivation of their name ar–arthropod, bo–borne, viruses. Transmission to humans (epizootic transmission) occurs by chance and is secondary to the natural enzootic cycle involving an arthropod vector and an avian or mammalian host. Mosquitoes serve as the vectors for the majority of clinically significant arbovirus endemic to North America. However, Colorado tick fever virus, Powassan (POW) virus in North America, and tick-borne encephalitis (TBE) virus in Eurasia are transmitted by ticks (214,215).
Flaviviridae
West Nile Virus
West Nile virus (WNV) was first isolated in Uganda in 1937 (216). Its incursion into the United States in 1999 (217) preceded a rapid spread throughout the contiguous continental United States as well as North and South America (218,219). In the United States, WNV has displaced all autochthonous arboviruses as the single major cause of CNS disease (220). WNV is a member of the Flaviviridae family of RNA viruses, within the genus Flavivirus (221
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


