TUBERCULOUS MENINGITIS
DOROTHEE HEEMSKERK, JEREMY FARRAR, AND MAXINE CAWS
Yea, I have known inflammations, Imposthumes, whelks, scirrhus Tumors growing to the Meninges, with the Skull, and other Diseases of an evil conformation, excited in the Membranes of the Brain; by which at first for a long time, frequent headache, and most cruel, and then afterwards a sleepy and deadly distemper hath been induced; the cause of the Disease not detected, but after Death by the Anatomy; and indeed it is to be suspected that inveterate and pertinacious pains in the Head, which return, and daily become more tormentive, in spight of all Remedies depend upon some such invincible Cause.
– Thomas Willis (1621–1675), from “De Anima Brutorum” (1672) (1)
HISTORY
Tuberculosis
Tuberculosis (TB) has been a part of everyday human life since ancient times. There is evidence of TB in man dating back to 4000 BC, but the disease may have been present even earlier. Before the discovery of the causative agent (Mycobacterium tuberculosis), TB, in its many forms, has had many different syndromal descriptions: phthisis, consumption, scrofula, Pott disease, or others less well known such as yaksma (Indian) and Chaky Onkay (Incan). The term “tubercle” was first used by Franciscus de la Boe, also known as Sylvius of Leyden (1614–1672). He stated that tubercles were often seen in the lungs of consumptives (2). He was also accredited with discovering a cleft in the brain consequently named the sylvian fissure, which we now know to be a preferential site for exudates formed in tuberculous meningitis (TBM).
The Origin of Tuberculous Meningitis
The earliest descriptions of intracranial TB date back to the seventeenth century. Physicians frequently used the term “acute hydrocephalus” or “dropsy of the brain” for a condition in children of which the etiology was unknown but presented with fever, headache, vomiting, and rapid death. Some of the historical descriptions vividly illustrate the despair of both patient and doctor; some pathologic descriptions were punctilious and very archetypal.
Nec minus a phlegmone et abcessu quam hujasmodi meningitis et tuberculis, cephalgiae lethales et incurabiles oriuntur (Sometimes the headaches, fatal and incurable, follow abscesses and swellings of the envelopes of the brain, as well as placques and tubercles of these membranes). (Willis, 1672 [1])
Willis was far ahead of his time as it was not until 150 years later that the tubercles found upon autopsy were regarded as a distinguishing aspect of the clinical syndrome, which was only then proposed to be tubercular meningitis.
Attempts were made to define this disease entity based on clinical and autopsy findings, but due to the multiform presentation both clinically and pathologically, consensus was not reached until Robert Whytt (1714 to 1766) lifted the disease out of obscurity with his treatise “Observations on the dropsy in the brain.” He gives a detailed account of 20 patients, dividing the disease in four different stages, according to the pulse of the patient. He emphasized that the terminology “Dropsy upon the brain” (ύδρω-κεφαλον [hydrocephalus] επι [on] τωενχεφαλω [the brain] as described by Hippocrates [460 to 377 BC] in his De Morbis Popularibus) is in these cases incorrect, because the accumulated fluid is not found between the skull and the dura mater but most frequently in the ventricles. The antediluvian technique proposed by Hippocrates to make a perforation in the upper part of the cranium to evacuate the fluid Whytt concluded was of no use (3). This publication gave an impetus to scientists to study this condition systematically and many contributions followed with different views on the origin of the disease. Some considered the arachnoid the seat of the pest, others lesions in the brain parenchyma, the ventricles, until finally Penn in 1825, who thought the origin was in the pia mater, called it meningitis (1).
The term tubercular meningitis was first used in 1836 by P.H. Green in the Lancet. Green introduced the term tubercular meningitis to describe the condition of the cerebral membranes, which were affected by tubercular lesions in nine tenths of the cases in a series of 45 children, who at the same time had tubercular deposits in the lungs or the abdomen. Green argued that these findings were a more essential characteristic of the disease than the accumulation of cerebrospinal fluid (CSF) (4,5). The condition was uniformly lethal.
Elucidating Pathogenesis
By the end of the nineteenth century, attempts were made to relieve symptoms from raised intracranial pressure and hydrocephalus. Walter Wynter (1860 to 1945) had devised a crude technique to puncture the lumbar subarachnoid space. He successively performed this archaic form of lumbar puncture on four patients with TBM to relieve symptoms but with short-lived improvement (widening of the pupils and temporary improvement of sensorium), but all four patients died (6,7). Morton (1891) describes the clinical findings of a series of patients during illness and the pathologic findings in the brain postmortem to further explore whether there would be a rationale for Wynter’s procedure. Although failing to clearly associate the extent of the hydrocephalus to the observed clinical picture, he concluded:
The operation does no harm, and as the patient is already comatose no anaesthetic is required. But in any efforts we may make to remove the more serious symptoms of tuberculous meningitis by draining the intraventricular fluid we must remember it is nearly always only part of general tuberculosis, which may, and probably will, prove fatal in other ways, though if in this rapidly fatal meningitis we can prolong life it may be some time longer before the general tuberculosis does its deadly work. (8)
Tuberculous meningitis remained universally fatal.
It was not Wynter but Heinrich Quincke who began to popularize the lumbar puncture for both therapeutic and diagnostic purposes in the late nineteenth and early twentieth century. With the discovery of M. tuberculosis by Robert Koch in 1892 and the development of x-rays by Wilhelm Roentgen in 1895, diagnosis before death was now achievable, although effective treatment remained elusive.
The disease was still thought to develop in a manner analogous to other meningitides until a meticulous serial autopsy study of 82 patients by Rich and McCordock (9) in 1933 provided the basis of what still now is considered to be the establishment of pathogenesis. In guinea pigs and rabbits, Rich and McCordock (9) could only provoke inflammation of the meninges by the direct inoculation of bacilli into the central nervous system (CNS) and not by peripheral injection and consequent hematogenous spread. In human autopsy studies, they described that the tubercles found in the brain were seldom of the same age as those found in other organs and that vasculitis found in the brain was rather a process originating from the adventitia inward, more likely to be the result of a focus within the brain rather than caused by direct hematogenous spread of bacilli. These findings led them to form a coherent hypothesis in which they postulated that, after inhalation of the pathogen, a short-lived bacteremia followed, during which bacilli spread throughout the body and seeded the surface of the brain, forming small granulomas known as Rich foci. These can exist without causing symptoms for an unknown period but may rupture; upon release of the mycobacteria, meninges become inflamed, giving rise to a multitude of possible pathologic tuberculous conditions in the CNS (9).
Treatment Development
The discovery of the causative pathogen was met with euphoria, as new hopes were raised of a cure. Experimental treatment was attempted with tuberculin, originally a glycerin extract of M. tuberculosis developed by Koch, but with catastrophic results. Other therapies raised hopes but proved ineffective, including sanocrysin (gold therapy) (10). In 1944, the first effective antituberculous agent, streptomycin, was discovered by Salman Waksman, a discovery for which he would win the Nobel Prize for Medicine in 1950. By 1948, dozens of cases of successfully treated TBM with intrathecal and intramuscular streptomycin were reported in the literature. Rich and Samuels (11) reviewed these cases while giving a striking account of a case involving a 2-year-old boy who at the height of his disease was in a vegetative state but within 6 months recovered with residual weakness of his left arm and minor mental impairment. Many publications followed on successful streptomycin treatment. However, by 1950, numerous streptomycin resistance reports appeared (12–17). Paraaminosalicylic acid (PAS) was added to the regimen; although a weak antituberculous agent, it prevented development of resistance. With the introduction of isoniazid (1952), a major improvement in treatment of all forms of TB was seen. It became clear that intrathecal administration was no longer necessary. With the introduction of pyrazinamide (1954) and rifampicin (1963), treatment regimens for all presentations of drug-susceptible TB could be shortened to 6 to 8 months. The relative contributions of the first-line TB drugs to the efficacy of TBM treatment were comprehensively reviewed by Donald in 2010 (18). Fifty years on these drugs isoniazid, pyrazinamide, rifampicin, and streptomycin remain the mainstay of treatment for the vast majority of patients with TB globally. It is difficult to think of another common infectious disease whose treatment regimen has remained largely unchanged for over 50 years.
Vaccine Development
Despite failing to find a cure, Koch continued experimenting with tuberculin hoping to develop an effective vaccine; however, it proved not to be effective. Clemens van Pirquet (1874 to 1929), after observing a hypersensitivity reaction to a second inoculation with smallpox vaccine, was led to the idea that Koch’s tuberculin might cause a similar reaction in patients previously exposed to mycobacteria. Charles Mantoux (1877 to 1947) expanded on these ideas and developed the Mantoux diagnostic test in 1907, which is still in use today. During 1902 to 1920, Albert Calmette and his assistant Camille Guérin developed a vaccine by serial passage of Mycobacterium bovis (which can cause TB in both cattle and humans). After 13 years and 230 passages, the bacille Calmette-Guérin (BCG) strain was considered attenuated and was first used as a vaccine in humans in 1921 (2).
The accomplishments in the first half of the twentieth century in diagnosis, treatment, and understanding of pathogenesis seemed to offer the opportunity of eradication. However, the next 60 years has been fraught with setbacks and TB remains a huge global public health problem. The first cases of AIDS came to light in the early 1980s. Since then, it has become clear that the interaction of TB and HIV has had a severe impact on both pandemics, while complicating the management of both diseases. HIV-infected individuals are more susceptible to both active TB disease and all extrapulmonary forms of TB. The HIV epidemic has therefore generated significant increases in the number of adults presenting with TBM in high HIV prevalence areas. Drug-resistant strains of M. tuberculosis have continued to increase in prevalence, including multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, which are now present in every region of the world. Huge advances have been made in unravelling immunology, and genetics has deepened our understanding of the immunopathology underlying disease; however, vaccination, diagnosis, and treatment are still reliant on antiquated techniques, which are inadequate to control the continued pandemic.
BURDEN OF DISEASE
Tuberculosis Epidemiology
In the nineteenth century, TB was highly prevalent in Europe, with an annual incidence estimated to be greater than 1,000 per 100,000. Mortality was high because patients were only treated with bed rest or ineffective therapies and it is thought that over 50% of sufferers died. In the time of Robert Koch, one out of seven Germans died of TB. In the late nineteenth and early twentieth century, incidence declined in more developed countries due to economic development, improved living conditions, hygiene, and the introduction of sanatoria in which patients were isolated from the general population. After the introduction of antimycobacterial treatment, the decline in incidence and mortality accelerated. The initial response was euphoric, and expectations to extinguish this blazing epidemic were high.
Despite the initial achievements during the twentieth century, global TB burden remains enormous. Worldwide, approximately 2.4 billion people are infected with M. tuberculosis, of whom 10% will develop active disease during their lifetime. People infected with HIV with latent TB are 20 to 30 times as likely to develop active forms of TB (19).
A systematic analysis for the global burden of disease study in 2010 including mortality data from 187 countries from 1980 to 2010 ranked TB as the 10th leading cause of death globally. Of the 52.8 million deaths of all causes globally in 2010, 1.2 million were attributable to TB (20). According to the World Health Organization (WHO), 8.7 million new cases were reported in 2011 and an estimated 1.4 million died (21). The Millennium Development Goals aimed to achieve a 50% reduction in prevalence and death rates of TB relative to 1990 levels by the year 2015. According to the WHO, from 1990 to 2011, a reduction of 41% in mortality was observed. According to the WHO, if the current trend is preserved, the set target for 2015 will be met (21). However, this is dependent on the continued commitment of national TB programs and policy makers. The TB epidemic is highly pluriform, with 22 developing countries carrying more than 80% of the burden of TB. Demographic factors such as poverty, crowding, and malnutrition play an important role, as does availability of good quality TB drugs. The impact of the HIV pandemic is illustrated by the huge increase in the contribution of HIV/TB to cause of death patterns among young adult men and women; by 2010, HIV/TB and injuries combined caused more than half of deaths among men aged 20 to 39 years (20). Together with HIV, MDR and XDR TB continue to fuel the pandemic (21). The goal to eliminate TB as a public health problem by 2050 seems quixotic without renewed and sustained commitment from international donors and the global health community.
Burden of Tuberculosis of the Central Nervous System
The burden of extrapulmonary TB is tightly associated with the general pandemic and accounts for more than 10% of all TB cases (21). Of these, about 5% are forms of TB in the CNS. These estimates are crude because diagnosis of extrapulmonary TB in general and CNS TB in particular is challenging, and according to WHO definitions, a patient who has both signs of pulmonary and extrapulmonary TB should be classified as having pulmonary TB (21).
In endemic settings, TB is often the leading cause of childhood bacterial meningitis (22). In 2009, 7% of all annual cases of bacterial meningitis and septicemia in the United Kingdom were caused by TB, being the third leading cause after meningococcal and pneumococcal disease (23). With the introduction of meningococcal C (1999) and pneumococcal (2006) vaccines in the routine immunization schedule in the United Kingdom (and in the absence of an effective TB vaccine), TBM may well become the lead cause.
In high TB-burden countries, young children aged 0 to 4 years are mostly affected, however, rarely, younger than 3 months of age (24). The age range from 5 to 15 years old is often referred to as “the favored age” as this population has the lowest rate of TB of all forms (25). In countries with a low prevalence of TB, most cases of TBM are in adults. Commonly reported risk factors are alcoholism, diabetes, recent corticosteroid use, malignancy, and immunosuppression (26). The advent of HIV has dramatically changed the dynamics of the TB pandemic.
Mortality and Morbidity
Mortality among TBM cases remains high and varies depending on age, risk group (HIV), stage of disease upon diagnosis, drug sensitivity of the infecting organism, time between onset of symptoms and initiation of effective antibiotics, and the sophistication of health care infrastructure and facilities. Mortality in children ranges between 10% and 20% (24,25,27,28). Neurologic sequelae are frequent and reported in more than half of surviving children. In a retrospective survey of 554 children in South Africa, 74% of patients suffered long-term disabilities or death (13%), including hearing or vision impairment (14% and 16% respectively), motor deficits (44%), and cognitive impairment (77%) (29).
For HIV-uninfected adult patients presenting early (stage I disease), mortality is approximately 20% (30). Mortality increases with delayed presentation and advanced stage of disease (Table 29.1). For patients in stage II, mortality is approximately 30% and in stage III, 55%. As in children, in adults, permanent neurologic disability affects over half of the survivors (30). Co-infection with HIV has a major impact on mortality. In Vietnam, mortality for the corresponding grade groups in a trial of immediate versus delayed antiretroviral therapy (ART) for HIV-associated adult TBM was stage I, 40%; stage II, 52%; and stage III, 75% (31). In HIV-infected children in India, 6-month mortality has also been reported to be drastically higher than in HIV-negative children (36% vs. 10%) (32). These mortality figures are a reflection of the severe immunosuppression in these HIV-infected patients, illustrating the importance of commitment of policymakers to invest in accessible integrated TB/HIV care.

The Influence of HIV
The HIV epidemic has undoubtedly fuelled the TB epidemic. Worldwide, an estimated 34 million people are infected with HIV (33). An estimated one third of these patients are thought to be co-infected with M. tuberculosis. HIV-infected patients with latent TB have a 50% lifetime risk of progressing to active forms of TB versus HIV-negative patients with latent TB who have a 10% lifetime risk of developing disease (34). HIV patients are more likely to develop extrapulmonary forms of TB, including TBM, and have a higher mortality. Diagnosis of TB can be more challenging in HIV patients due to a less specific presentation, wider differential diagnosis, and lower sensitivity of sputum smear microscopy. However, in contrast, CSF smear is more likely to be positive in HIV-infected individuals with TBM due to higher bacillary loads (35). Patients with low CD4 levels and smear-negative results may have atypical CSF findings, immunologic tests are not reliable, and neuroimaging may not reveal typical lesions and includes a wider differential diagnosis (36). Polypharmacy may prove problematic, with higher toxicity from combined ART and antituberculous regimens and drug interactions. ART-naive patients who commence ART treatment during their treatment of TBM may present with immune reconstitution inflammatory syndrome (IRIS), which may be particularly detrimental if it presents intracranially (37).
Drug Resistance
WHO estimates 20% of M .tuberculosis infections worldwide are now resistant to at least one first-line drug (38). MDR TBM is resistant to at least rifampicin and isoniazid, the two most effective first-line agents. MDR TBM has been associated with very high mortality in both children and adults (39–41). Drug resistance in TBM is rarely diagnosed in time to make appropriate treatment adjustments. The difficulties of access to rapid TB drug susceptibility testing (DST) in much of the world are compounded by the rarity of a positive isolation of M. tuberculosis from the CSF. Drug resistance prevalence among TBM cases will generally follow similar patterns to those observed regionally for pulmonary TB.
Isoniazid resistance in the absence of concomitant rifampicin resistance is more prevalent than MDR TB; 7% of M. tuberculosis strains globally are now resistant to isoniazid (38). Because isoniazid is the most effective drug in decreasing the bacterial load in the first two days of anti-TB treatment, early recognition of resistance is of great importance, particularly in TBM cases where rapid killing of bacilli is likely to be crucial. In Vietnam, isoniazid resistance alone or combined with resistance to other drugs was found in a third of culture-positive samples in adult HIV patients with TBM, with 4.3% MDR TBM (40). In a cohort with predominantly HIV-negative patients, isoniazid resistance was found in 37.1% of samples of which 21% were MDR (overall MDR rate was 5.6%) (39). Mortality from MDR TBM was 100% in the absence of available second-line therapy. In children with a culture-proven diagnosis of TBM, MDR TB or rifampicin monoresistance was identified in 5% of cases in South Africa. Multidrug resistance, not surprisingly, was associated with very high mortality (83%) (42).
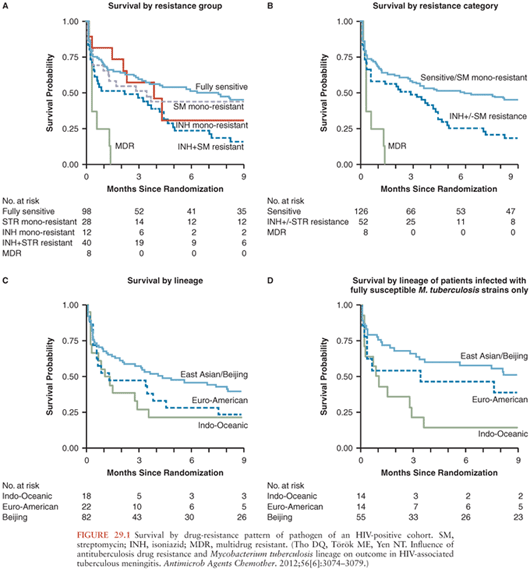
Isoniazid resistance without rifampicin resistance also has a significant impact on mortality. An initial analysis of a cohort of TBM patients in Vietnam failed to find a significant impact of isoniazid resistance (+/− streptomycin resistance) on mortality (43), but a larger study in Vietnamese HIV-positive patients showed a significant reduction in survival (adjusted hazard ratio [HR], 1.78; 95% confidence interval [CI], 1.18 to 2.66) among patients infected with isoniazid-resistant, rifampicin-susceptible strains compared to those with fully susceptible isolates (40). In a retrospective cohort study in the United States, researchers also found a significant increase in risk of death associated with isoniazid resistance (odds ratio [OR], 1.61; 95% CI, 1.08 to 2.40) (44). Among Vietnamese HIV-positive adult patients, the adjusted hazard ratio for mortality of MDR TBM patients compared to patients with isolates susceptible to all agents or streptomycin monoresistant was 5.21 (95% CI, 2.38 to 11.42) (Fig. 29.1).
Long-Term Disability
Very little is known about long-term outcome and disability for both children and adults. Antituberculous chemotherapy has been unsuccessful in completely preventing long-term sequelae; in many cases, diagnosis may be too late, but in others, significant neurologic damage occurs subsequent to initiation of treatment. Especially in children, neurocognitive impairment can jeopardize development, education, and quality of life and place a great burden on families, schooling, and medical systems. A recent long-term follow-up study on a South African cohort of pediatric TBM patients who were severely ill on presentation (stage II or III) reported only 20% of children to be functionally normal at follow-up (median 6 years after TBM treatment completion). The main areas of functional deficit were cognitive impairment (80%), poor scholastic progress (43%), and emotional disturbance (40%). A smaller proportion of children (25%) had evidence of motor impairment (45). In a Danish nationwide, population-based cohort study with up to 30 years follow-up, TBM was associated with an almost twofold increased long- term risk of dying compared to a background population (mortality risk ratio [MRR], 1.79; 95%, CI, 1.09 to 2.95). In this study, the underlying cause of long-term death was most frequently TBM itself rather than secondary to the most commonly reported neurologic sequelae (46).
Neurologic sequelae most frequently described in adults are cognitive impairment, motor deficits, cranial nerve palsy, and optic atrophy (47). A 5-year follow-up on Vietnamese adults with TBM who took part in a randomized controlled trial on the effect of dexamethasone on survival could only demonstrate an overall benefit of dexamethasone up to 2 years following treatment. Five years after treatment completion, there was no difference in overall survival or disability outcome in both groups. In the group receiving dexamethasone, 48.4% (vs. 52.7% in placebo group) of patients had died at 5 years (31% at 9 months), 6.8% (vs. 7.4% in the placebo group) were severely disabled, 17.2% (vs. 14.8% in placebo group) had intermediate disability, and only 27.6% (vs. 25.1% in the placebo group) had good outcome (48). However, the beneficial effect of dexamethasone was preserved at 5 years for patients with stage I TBM at presentation, demonstrating that, contrary to preceding medical wisdom, patients with stage I TBM are the group who gain the greatest benefit from corticosteroids.
Vaccination with Bacilli Calmette-Guerin and Protection Against Tuberculous Meningitis
The controversies surrounding the protection that the BCG vaccine confers to adult pulmonary and meningeal TB still exist. In adults, reported efficacy against all forms has ranged from as high as 80% to 0%. It is proposed that the vaccine establishes immunity by inducing effector memory T cells in the lungs that gradually wane after 10 to 15 years rather than building a longer lasting “central memory” (49). Various theories have been proposed for the differences observed in efficacy, including differences in the circulating M. tuberculosis strains, BCG vaccine strains, or preimmunization exposure to environmental mycobacteria. Nonetheless, consensus on the benefit of prevention of severe forms of childhood TB including TBM and miliary TB is established. The vaccine is thought to be 52% to 86% protective against developing the severe complications of childhood TB such as miliary TB and TBM (50). It has been estimated that the 100.5 million BCG vaccinations given to infants in 2002 would have prevented 29,729 cases of TBM in children during their first 5 years of life, or 1 case for every 3,435 vaccinations, and 11,486 cases of miliary TB, or 1 case for every 9,314 vaccinations. Based on these data, it is considered a cost-effective intervention in Southeast Asia, Africa, and the Western Pacific, where TB infection rate and vaccine coverage are highest (50). In BCG-vaccinated children in India who do develop TBM, the clinical spectrum of disease does not seem to be ameliorated (51). The BCG is the most widely used vaccination globally.
IMMUNOPATHOGENESIS
Mycobacterium tuberculosis
Transmission of TB occurs when a person inhales mycobacteria-laden droplet nuclei. One to five bacilli are sufficient to cause infection. M. tuberculosis is the causative agent of almost all cases of TBM. M. tuberculosis is an obligate aerobic, intracellular bacterium. On the cell surface, it has a waxy coating. The high lipid content of the cell wall renders these bacilli imperceptible to the Gram stain. The organisms are slow growing with a generation time of 15 to 20 hours, contrasting with that of some pyogenic bacteria, such as Streptococcus pneumoniae, Neisseria meningitides, and Staphylococcus aureus, with generation times of less than an hour. The complex antigenic structure of the cell wall includes polysaccharides, proteins, peptides, lipids, and glycolipids with specific immunologic properties. Other antigens are contained within the cytoplasm. These molecules determine the characteristic immune response to tuberculous infection and its resultant pathology.
Macrophages and Granuloma Formation
In pulmonary TB, the alveolar macrophage has a central role in the initial innate immune response to M. tuberculosis as well as in initiating the adaptive T-cell immunity. If upon recognition and ingestion of bacteria, the macrophages fail to eradicate M. tuberculosis, T cells are recruited to the site of infection and generate a chronic inflammation and granuloma formation in order to contain the infection (52). The granuloma is pivotal to tuberculous disease and characterized by the formation of central necrosis of the lesion, often referred to as caseation. In solid necrosis, mycobacterial growth is inhibited, and the infection can be contained and can remain dormant. Granuloma with liquefied central necrosis provides an optimal environment for extracellular mycobacteria and may rupture, allowing the spread of bacilli to other parts of the lung, the bloodstream, or the exterior environment (53).
The onset of the adaptive immune response to M. tuberculosis is delayed compared to other infections. This delay allows for an exponential growth of the mycobacteria before it is slowed or arrested by the host defenses (54). For the adaptive immune response in TB, CD4+ T cells are essential. CD4+ T cells exert their protective effect by the production of cytokines, primarily interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). IFN-γ is a central protective cytokine in mycobacterial infection proposed to protect by mediating the induction of nitric oxide synthase (NOS), enhancing the microbicidal system within macrophages (55). TNF-α plays a key role in granuloma formation and macrophage induction and has immunoregulatory properties (53). Other cytokines involved in mycobacterial disease control are interleukin (IL)-1B, IL-6, IL-12, IL-15, IL-18 (proinflammatory) and IL-10, IL-4, and transforming growth factor-β (TGF-β) (antiinflammatory) (53,56). In addition to the complex regulatory meshwork of cells and immune mediators that control the innate and adaptive immune response, different components may be negatively influenced by microbe-specific virulence factors. The virulence of the mycobacteria is thought be founded in their ability to regulate macrophage chemotaxis, necrosis, and apoptosis, facilitating a beneficial environment within the macrophage or extracellularly in the granuloma and consequently their own proliferation and possibly also facilitating egress to other sites of infection, including the CNS (57).
Immune Response in Tuberculous Meningitis
TNF-α is thought to have a crucial but controversial role in pathogenesis of TBM. TNF-α is a proinflammatory cytokine produced by monocytes, macrophages, and dendritic cells upon stimulation with mycobacteria or mycobacterial products and plays an essential role in granuloma formation and maintenance (58). Whereas in pulmonary TB, TNF-α-neutralizing drugs can lead to progression to (fatal) disease, in TBM, an elevated production of TNF-α has been proposed to be associated with more severe disease, but it has not been established if this is merely a marker of advanced disease or if disproportionate TNF-α responses mediate disease progression. It is probable that protective immunity is dependent on a delicate balance between pro- and antiinflammatory factors and that individuals with responses at the extremities of the spectrum are both at risk of more severe disease.
In vitro infection of microglial cells (macrophages of the brain) results in the production of robust amounts of TNF-α, IL-6, IL-1B, CCL2, CCL5, and CXCL10 (59). In rabbits infected intracisternally with M. bovis, oral thalidomide treatment led to reduction of TNF-α levels and clinical improvement (60). In vivo in TBM, TNF-α levels show a peak in the early phase of disease but drop soon after initiation of treatment (61). Some researchers found an association in disease severity and CSF levels of IFN-γ and TNF-α in both HIV-positive and HIV-negative patients (62). This was not supported in a Vietnamese cohort, in which the only cytokine independently associated with severe disease was IL-6. HIV co-infection was associated with attenuated levels of several immune mediators in CSF, in whom low levels of IFN-γ did show an association with death, implying a protective role for IFN-γ. Interestingly, the addition of dexamethasone to treatment was not associated with an attenuation of inflammatory indices but did lead to a decreased mortality rate (61).
The brain provides a unique immunologic environment to pathogens. We know most of the inflammatory mediators are produced locally at the site of infection. Immunologic studies usually involve cells from peripheral blood or CSF. Compared to the infected tissue, even in pulmonary TB, low proportions of M. tuberculosis–specific effector cells are found in the blood (63).
Central Nervous System Pathology
Unfortunately, current postmortem studies are scant (64,65) but would contribute a great deal to our understanding of the disease. Rabbit models are thought to mimic human disease; however, immunologic interpretation is imperfect. Murine studies are not of use in directly studying neuropathogenesis in TBM although are widely used in understanding susceptibility and protective responses to mycobacteria (66). Our current knowledge on pathogenesis of CNS TB is based on the hypothesis promulgated over 70 years ago. Even though later researchers have expressed criticism (67), their view has been carried forward by most recent experts; in short, small intracranial granulomas are formed after seeding of bacilli in the brain during a short-lived bacteremia. These Rich foci may rupture during periods of relative immunosuppression or other unknown stimuli, releasing mycobacteria in the subarachnoid space or ventricular space which may give rise to the various forms of CNS TB: meningitis, tuberculoma, tuberculous abscess, encephalitis, or spinal cord TB. Incorporating more recent pathologic data, Donald and colleagues (68) have proposed the following classification of pathogenesis in TBM:
1. The hematogenous dissemination of bacilli from the primary complex establishes a cortical or meningeal focus. Soon after its establishment, this proceeds to caseate and discharge its contents into the subarachnoid space. In young children, this hematogenous dissemination is particularly likely to take the form of miliary TB.
2. In a small minority of cases, hematogenous dissemination may establish a caseating focus in the choroid plexus or in the walls of the ventricles from which TBM may develop.
3. Hematogenous dissemination at the time of primary infection, or later, establishes a cortical or meningeal focus. This is initially controlled but may, at any time thereafter, undergo caseation and discharge its contents into the subarachnoid space.
4. A caseous process extends from adjacent structures such as the vertebrae or middle ear to involve the CNS (very rare).
As a result of this infection, a dense gelatinous fibrinocellular leptomeningeal exudate is formed. Microscopically, this exudate contains small and large mononuclear cells, including epithelioid cells, which also act as macrophages. The exudate typically centers around the interpeduncular fossa. When substantial, the exudate may extend anteriorly to the suprasellar region, and it may extend through the prepontine cistern and surround the spinal cord and cerebellum, often into the sylvian fissures. It can envelope and compress cranial nerves and arteries. Vasculitis may develop, giving rise to ischemic events. Hydrocephalus can develop by blockage of CSF circulation when exudates cover the choroid plexus and the basal subarachnoid cisterns around the midbrain and pons or when tuberculoma cause narrowing of the aqueduct and third ventricle (66,69). Exudate, vasculitis, and hydrocephalus can cause changes in brain parenchyma. “Border-zone encephalitis” describes a tissue reaction commonly seen in brain tissue adjacent to zones of thick adherent exudate. The brain tissue softens, showing signs of edema, perivascular infiltration, and microglial reaction (66,70). In the following paragraphs, we will briefly discuss other forms of CNS TB; the rest of the chapter will focus on TBM.
Tuberculoma in the Central Nervous System
CNS tuberculoma can be encountered separate from TBM, as it is estimated only 10% of patients with tuberculoma develop meningitis (71,72). Conversely, in some radiologic studies, tuberculomas were observed either at presentation or developed during treatment in over 60% of patients with TBM (73). In endemic areas, tuberculoma represents the cause of up to 30% of patients presenting with intracranial masses. Tuberculoma can be solitary or multiple, with some reporting hundreds of lesions in one patient, dubbed “tuberculomatosis cerebri” (71). In general, tuberculomas in the context of TBM, although depending on the location, are not associated with worse outcome and will resolve on antituberculous treatment (74). Rarely, tuberculomas coalesce and liquefy to cause tuberculous cerebral abscess, which may necessitate surgery (75).
Tuberculous Encephalopathy
Tuberculous encephalopathy (TBE) was first described in 1966 in Indian children who presented with symptoms of a diffuse cerebral involvement (coma, convulsions, movement disorders) in the context of disseminated TB but normal cerebrospinal findings (76). As there was no clear evidence of TB infection within the brain, the authors proposed an alternative pathogenetic immune-/hypersensitivity-mediated explanation for this syndrome, pathologically characterized by white matter, myelin loss with commensurate axonal loss, and focal necrosis. The principal pathogenetic factor in the group of cases was said to be an allergic cerebral edema leading to an edematous leukoencephalopathy similar to acute disseminated encephalomyelitis (ADEM) (77). However, use of steroids for these patients had proven ineffective. Careful review of the original publication and the literature of the following 40 years led South African experts to reappraise this entity in 2007, concluding that the patient population joined under the umbrella of TBE was clinically and histopathologically heterogenous. According to the authors, other plausible factors may have caused the typical findings of TBE, including hypoxic ischemia and toxic or drug-related complications of TB infection (78). TBE has not been reported in adults.
Tuberculosis of the Spinal Cord
Tuberculous radiculomyelitis (TBRM) has been used as a generic term to include arachnoiditis, intramedullary tuberculoma, and spinal cord complications of TBM (79). Currently, it is thought that TBRM may develop in alternative ways, either (1) as a primary lesion, (2) as an extension from TBM, or (3) secondary to vertebral TB (80). Intramedullary tuberculomas are rare. The pathogenesis is parallel to CNS TB: via bloodborne seeding of bacilli, granuloma formation follows, drainage persists into the subarachnoid space which may lead to an inflammatory reaction in the pia-arachnoid, which may lead to secondary meningitis (81). Clinical presentation may vary, usually presenting with symptoms of a subacute intramedullary space-occupying lesion (82). Early recognition of this form of CNS tuberculoma is important because early surgical intervention and decompression followed by long-term antituberculous chemotherapy may significantly improve morbidity. Accurate diagnosis can be helped by magnetic resonance imaging (MRI) (83).
Vascular Events
Secondary to the infection with M. tuberculosis and resultant intracerebral immunologic response, stroke may develop. In the late nineteenth and early twentieth century, vascular involvement in TBM was recognized and extensively studied. Still the vascular events in TBM are a pressing subject in TBM research, because the damage caused by stroke is often irreversible and the occurrence of stroke is associated with worse outcome (84,85).
Stroke was reported in 13% to 57% of Indian patients with TBM (85). In Vietnam, serial MRI revealed stroke in only 9% of patients upon diagnosis, but after 60 days of treatment, 41% of patients had developed stroke (73). This may be even more prevalent in children, with infarcts reported in 76% of systematically scanned children in South Africa (86). Most infarcts occur in the region of the arteria cerebri media (middle cerebral artery [MCA]), particularly in the medial lenticulostriate and thalamoperforating vessels, causing the characteristic basal ganglia infarcts (84). It is thought that this vascular involvement follows the distribution of the meningeal exudates, which causes local vasculitis, particularly at the base of the brain and along the sylvian fissures. The proposed mechanisms by which the vessels occlude resulting in ischemia are multiform. Meticulous histopathologic descriptions of the vascular changes in TBM were published by Hektoen (87) in the late nineteenth century. He concluded that the changes could be either explained by an endarteritis with subendothelial tubercles, proposed to be caused by direct hematogenous invasion of bacilli in the vessel wall, or tuberculous proliferation affected the arteries from the adventitia inward to reach the media and the intima (87). Currently, largely based on the findings of Rich and McCordock (9), the widely accepted view is that the inflammation is spread from outward (adventitia) in rather than reversed. Other stenosing or damaging mechanisms are thought to be intimal proliferation, vessel wall necrosis, or vasospasms. The role of vessel thrombosis is unclear. There is some evidence that strokes early in the course of disease are caused by vasospasms and later strokes involve proliferative intimal disease, raising the prospect for therapeutic interventions for the prevention of thrombotic and vasospasm-associated stroke (9,84).
The Role of Miliary Tuberculosis
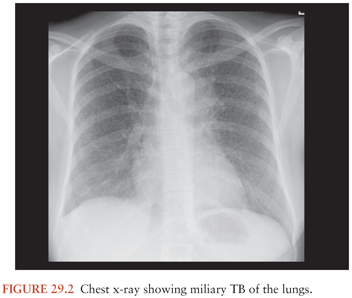
Rich and McCordock (9) did not assign a role for miliary TB in the pathogenesis of TBM as the Rich foci found in their subjects were often older than the miliary lesions, and therefore military TB was deemed to be an incidental occurrence rather than part of the etiology. Even though this is the view carried forward in most textbooks, some researchers claim that, particularly in children and possibly in immunocompromised patients, the disseminated state of miliary TB increases the likelihood of the formation of leptomeningeal granuloma (Rich foci) (68). This may well explain the frequent association of TBM with miliary TB (Fig. 29.2). In children, the prevalence of miliary TB of the leptomeninges is much higher than in adults. Younger children most often develop both TBM and miliary TB within 3 months of primary infection. Children with concomitant miliary TB and TBM are also significantly younger than those with TBM only (24). The immune system of these young children and the immunocompromised may not be robust enough to prevent an overwhelming bacteremia, exemplified by the miliary character of disease. In these individuals, TBM may be the result of a more direct spread of the pathogen to the meninges and subarachnoid space.
CLINICAL PRESENTATION
General Symptoms on Presentation
TBM typically presents in a subacute manner. Presenting signs, symptoms, CSF findings, and frequencies according to the British Infection Society guidelines are shown in Table 29.2. In adults, the majority of patients present with fever, headache, nuchal rigidity, vomiting, meningism, abnormal mental stage, and photophobia (75,88,89). Weight loss, night sweats, lethargy, and cough have also be reported (90). The mean duration of symptoms is typically more than 5 days. A longer duration of history is associated with worse symptomatology on presentation.
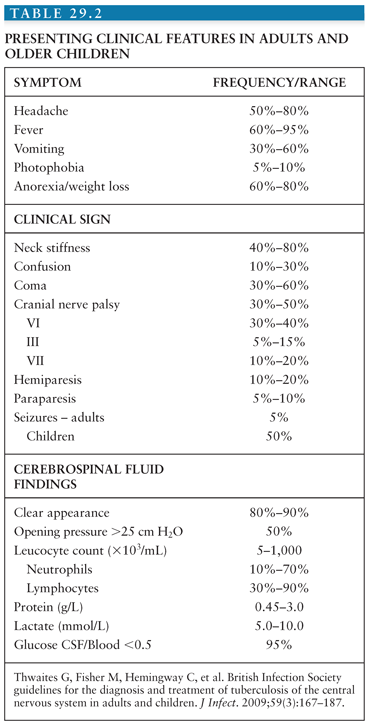
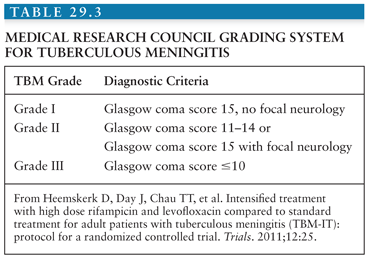
In 1948, the British Medical Research Council first published a classification of TBM patients according to the severity of disease (see Table 29.1). Over the years, these stage groups have been refined and are published in different formats. In general, both adult and pediatric patients in stage I are fully conscious and may have nonspecific symptoms; in stage II, patients will have signs of meningitis, lethargy, or cranial nerve palsies; in stage III, TBM is accompanied by stupor, severe illness, gross paralysis, or paresis, convulsions, and or involuntary movements (51). In clinical practice and research, it would be more useful to have a Glasgow Coma Scale (GCS)–guided staging system. An example is shown in Table 29.3.
Typical findings upon neurologic examination are VI cerebral nerve palsy (present in up to 40% of patients) but also nerves III and VII are often involved (5% to 20%). Hemiparesis and paraparesis may be present upon presentation (in 5% to 20% of patients) (75,88) but may also develop during treatment secondary to infarction. Seizures are rarely a presenting symptom in adults, however in children are reported in around 50% of patients (28,29).Visual disturbances, decreased vision, and diplopia may have a variety of causes, including primary involvement of optic nerve by tuberculous lesion leading to optic neuritis, optochiasmatic arachnoiditis (OCA), and tuberculoma in the chiasmatic region or in the optic pathways (91). More often, visual disturbances are secondary to raised intracranial pressure or ethambutol toxicity. A rare cause of visual loss in TBM is neuroretinitis (92). Tuberculoma can cause a wide array of neurologic symptoms associated with space-occupying lesions depending on their location within the CNS. Urinary retention is common which may indicate spinal cord involvement. Movement disorders are associated with basal ganglia involvement, mostly tremor, but also chorea, ballismus, and myoclonus have been reported (88).
The initial symptoms of TBM may be nonspecific, but within the context of prolonged symptoms, a previous history of TB, or a chest x-ray consistent with recent or past TB infection, this history must raise heightened suspicion with the treating physician. In many textbooks, TBM is described as chronic or subacute meningitis; however, this terminology is unhelpful. Once a patient with TBM seeks medical care, he or she should be treated as a medical emergency as with any other meningitis.
Children may have a more protracted and nonspecific presentation. In TB-endemic settings, it is often the most common cause of childhood bacterial forms of meningitis (22). Prodromal symptoms include fever, headache, anorexia, and vomiting in older children, whereas failure to thrive, poor appetite, vomiting, and sleep disturbances are more common in younger children or infants. Cough and weakness may also be reported (28,29).As these symptoms are nonspecific, children tend to present to the hospital only when the clinical situation has deteriorated and they are already in the later stages of disease (29). Similar to adults, when initial nonspecific symptoms are associated with a history of recent contact with a case of documented TB, TBM should be suspected. A prolonged history of more than 5 days, focal neurologic deficit, and abnormal movements have been found to be independently predictive of TBM (27). Examination may reveal nonspecific signs of meningism, failure to thrive, and in younger children, a bulging fontanel or increased head circumference. Funduscopy may reveal signs of papilledema or retinal involvement. In a retrospective study of 554 South African children, 97% presented in stage II or III, with a median duration of symptoms of 9 days. Meningeal irritation was the most frequent finding (98%). Convulsions (47%) and mono-/para-/quadri-/hemiplegia (63%) were also frequently observed. Cranial nerve palsies were less common (27%) than in adults, as was raised intracranial pressure (23%) (29).
The emergence of HIV has changed the epidemiology of TB and particularly the clinical outcome of disease. HIV-infected patients are more likely to develop extrapulmonary forms of the disease. TBM is considered an AIDS-defining condition. Research studies do not suggest that HIV greatly alters the clinical presentation of TBM, especially in patients with higher CD4 counts; the clinical presentation may mirror that seen in HIV-negative individuals. Patients with lower CD4 counts may have a more atypical course of disease, with less specific and more subtle signs and wider differential diagnosis, rendering diagnosis more challenging (36). Therefore, HIV-infected patients may present later during the course of disease, with altered consciousness, and consequently more often in the advanced stages of disease (35). HIV patients are more likely to have impaired cognition, generalized lymphadenopathy, and hepatosplenomegaly (32). A retrospective study comparing clinical presentation and outcome in children with and without HIV infection surprisingly found that HIV-uninfected children were more likely to present with a decreased level of consciousness; this may be related to the poor immune response in immunocompromised children. Similar to adults, HIV-infected children had a longer history of being unwell, poorer nutritional state, more commonly had accompanying hepatosplenomegaly, lymphadenopathy, and otorrhea (93). Despite the similarity in presentation, outcome is significantly worse for HIV-infected adults and children with TBM.
Progression During Treatment
Paradoxical responses during antituberculous treatment are frequently reported despite appropriate chemotherapy with susceptible bacilli. This can be encountered in all tissues but most often in the lungs, lymph nodes, and the brain (94). In the brain, tuberculoma may develop or enlarge during treatment for pulmonary TB, TBM, or miliary TB. Either discovered on routine brain imaging or accompanied by worsening of symptoms, signs of a space-occupying lesion, or convulsions. This generally occurs within 1 to 4 months of starting treatment, often after initial improvement. Antituberculous therapy should be continued; the addition of systemic corticosteroids may be considered (95). Within the context of a sound diagnosis and microbiologic confirmation of a susceptible pathogen, paradoxical response can be diagnosed clinically; however, incorrect diagnosis, drug resistance, and cerebral infarction may be alternative causes of deterioration despite treatment. Early recognition of these alternative causes is important as they warrant urgent intervention.
In TBM, vascular events are most often ischemic in nature. Vascular involvement is more frequently seen in chronic meningitides than in treated acute bacterial forms of meningitis (96). Other infective causes of stroke in tropical regions may include malaria, syphilis, Chagas disease, cysticercosis. Events can go unnoticed, as they occur silently or in severely ill patients already in deep coma. The most common signs of TBM-associated stroke are mono- or hemiplegia but also may present as lowered consciousness, disorders of movement, seizures, cranial nerve palsies, papilledema, and decerebration (84). Clearly, neurologic deterioration also can have its origin in a tuberculoma, cerebral edema, or infiltrating exudate (97). Unlike hypertensive or atherothrombotic stroke, transient ischemic attacks (TIAs) and lacunar lesions are rare in TBM (84). Patients may present with symptoms of stroke but more often develop stroke later in the course of disease, typically during the first weeks of treatment (73,84). It is not currently possible to predict which patients will develop stroke and it is associated with higher mortality and morbidity. Initial imaging studies may not be sensitive enough to detect the early changes of an ischemic event. Antithrombotic therapies such as aspirin and dipyridamole which prevent or reduce the incidence of stroke may improve outcomes in TBM and warrant further clinical study.
DIAGNOSIS
Clinical Case Definition
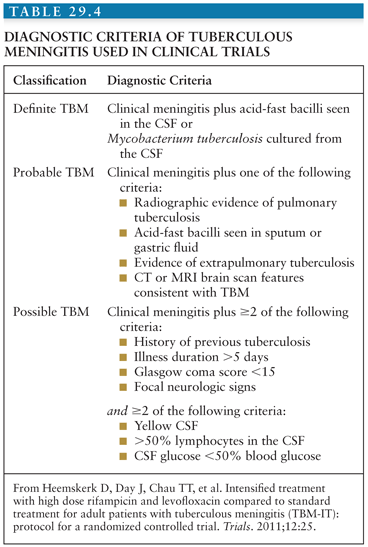
Early recognition of TBM is pivotal, because prompt initiation of treatment greatly increases chances of survival and reduces disability. However, early symptoms are nonspecific and diagnostic confirmation has hardly improved since the early twentieth century. Ziehl-Neelsen smear for acid-fast bacilli is central to diagnosis because it gives rapid results; however, the reported sensitivity is low. Sensitivity estimates depend on the criteria used for gold standard and range widely from 10% to 60%. This wide variation is likely to depend on many factors including laboratory performance, workload, technician diligence and experience, time from taking the sample to staining in the laboratory, and volume of CSF examined. Liquid culture of M. tuberculosis is considered the gold standard for diagnosis, but due to the slow growing nature of mycobacteria, the time to a positive result may range from 2 to 8 weeks. This renders the test ineffective for clinical decision making regarding treatment initiation, although a positive result can confirm the decision to continue therapy (although a negative result should not automatically lead to stopping) and provides an isolate for drug susceptibility evaluations. A suggestive history must raise clinical suspicion. A clinical diagnostic algorithm based on prospective data and validated against a second data set is available based on clinical and laboratory features (98,99). Different clinical algorithms are published throughout the literature; those of most use are based on simple clinical and laboratory criteria and can be used in resource-limited settings, where disease burden is highest. Table 29.4 shows an example.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


