The term thrombosis refers to the formation, from constituents of the blood, of a mass within the venous or arterial vasculature of a living animal. Hemostatic thromboses, namely, self-limited and localized thromboses that prevent excessive blood loss, represent the body’s natural and desired response to acute vascular injury. Pathologic thromboses such as deep venous thrombosis (DVT), pulmonary embolism (PE), coronary arterial thrombosis leading to myocardial infarction (MI), and cerebrovascular thrombotic occlusion represent the body’s undesired response to acute and chronic perturbations of the vasculature, blood, or both. The terms coagulated blood and clot are not synonymous with thrombosis and refer to the formation of a solid mass of blood components outside of the vascular tree. Examples of clots include soft tissue, body cavity (e.g., peritoneal), and visceral hematomas. Coagulated blood best describes clots formed ex vivo.
Thrombosis of the veins and arteries, together with complicating embolic phenomena, is perhaps the most important cause of sickness and death in the developed countries of the world at the present time. Deaths from MI and thrombotic stroke consistently represent the major causes of death in the United States, numbering >700,000 people annually (almost 30% of all deaths) in a recent report.1 Venous thromboembolic disease is the third most common cardiovascular disease, after atherosclerotic heart disease and stroke. It has been estimated that between 500,000 and 2 million venous thromboembolic events (VTE) including calf vein thrombosis, proximal DVT (e.g., lower extremity and pelvic veins), and PE occur annually in the United States alone.2 The incidence of pathologic thrombosis, prevalence of disorders that predispose to thrombosis, and morbidity and mortality associated with thrombotic events all reflect the magnitude of the importance of the study of thrombotic mechanisms and development of effective antithrombotic and thrombolytic therapies.
In this chapter, the pathophysiology of arterial and venous thrombosis and mechanisms of action of antithrombotic pharmacologic agents including antiplatelet drugs, anticoagulants, and fibrinolytic agents are summarized and discussed. Inherited conditions that predispose an individual to thrombosis, also termed hypercoagulable states, and the management of venous thromboembolic disease are covered. The management of arterial thrombotic events is beyond the scope of this chapter, and the reader is referred to several excellent recent review articles.3, 4, 5
PHYSIOLOGY AND PATHOPHYSIOLOGY OF THROMBOSIS
The human hemostatic system consists of multiple independent, yet integrally related, cellular and protein components that function to maintain blood fluidity under normal conditions and promote localized, temporary thrombus (hemostatic thrombus) formation at sites of vascular injury. A normal hemostatic system is the human physiologic defense against exsanguination. An abnormal hemostatic system can result in pathologic bleeding, vascular thrombosis, or both.
The hemostatic system is comprised of six major components: Platelets, vascular endothelium, procoagulant plasma protein “factors,” natural anticoagulant proteins, fibrinolytic proteins, and antifibrinolytic proteins. Each of these six hemostatic components must be present in fully functional form, in adequate quantity, and at the proper location to prevent excessive blood loss after vascular trauma and, at the same time, to prevent pathologic thrombosis. The hemostatic system is highly regulated and maintains a delicate balance between a prohemorrhagic state and a prothrombotic state. Any significant acquired or congenital imbalance in the hemostatic “scales” can lead to a pathologic outcome.
Normal hemostasis in response to vascular injury can be divided into two major processes of equal importance, known as primary and secondary hemostasis. Primary hemostasis comprises the reactions needed to form a platelet plug at a site of vascular damage, whereas secondary hemostasis comprises a series of reactions (coagulation cascade) needed to generate cross-linked fibrin required to stabilize the platelet plug and form a durable thrombus. Natural anticoagulants (antithrombin [AT] and activated protein C [APC]) function to confine thrombus formation to the sites of vascular injury and limit thrombus size to prevent vessel occlusion and flow interruption in the affected vessel. The activity of AT is greatly enhanced by endothelial cell heparan sulfate and pharmacologic heparins. The function of APC is enhanced by its cofactor, protein S. Physiologic fibrinolysis is initiated by endothelial-cell-derived tissue-type plasminogen activator (t-PA), which converts plasminogen to plasmin. Plasmin can degrade cross-linked fibrin, limit thrombus size, and help dissolve a thrombus once the vascular injury has been repaired. The fibrinolytic system is regulated and localized by antiplasmin and PA inhibitor (PAI)-1. Details of the hemostatic mechanisms and endothelial cell regulation of hemostasis are given in Chapters 18 and 19.
Specific alterations in the quantitative and qualitative status of any hemostatic cellular or protein element can lead to pathologic thrombosis. A marked increase in the platelet count (thrombocytosis) and accentuated platelet aggregation (“sticky platelet syndrome”) are associated with thromboembolic events. Elevated levels of procoagulant factors such as fibrinogen, factors VIII, IX, XI, and VII, as well as resistance to inactivation of factor Va by APC, are recognized risk factors for vascular disease and thrombosis. Deficiency of a natural anticoagulant protein such as protein C, protein S, or AT is associated with venous thromboembolic disease. Deficiency of a fibrinolytic cascade component, such as t-PA or plasminogen, and excess plasma levels of the fibrinolytic inhibitor PAI-1 have been linked to hypercoagulability and thrombosis. It is the net balance between the participating and, at times, opposing groups of proteins and not the level of any individual factor that is most critical to hemostatic regulation.
VIRCHOW TRIAD
In the mid-19th century (1854), German pathologist Rudolph Virchow postulated that vascular obstruction was precipitated by, and thrombosis resulted from, three interrelated factors: (a) “Decreased blood flow” (stasis of blood flow), (b) “inflammation of or near the blood vessels” (vascular endothelial injury), and (c) “intrinsic alterations in the nature of the blood itself” (hypercoagulability).6 Many students of coagulation medicine, though, do not realize that Virchow actually recognized his “triad” as being the result of vascular occlusion, not necessarily as the original precipitant of vascular thrombosis. Nonetheless, the vascular, rheologic, and hematologic aspects of thrombosis known as the Virchow triad remain relevant and instructive today (Fig. 55.1).
Abnormalities of Blood Flow
Arterial thrombosis initially occurs under conditions of rapid blood flow (high shear stress), a condition in which von Willebrand factor (vWF) is critical for platelet adhesion.7 Arterial thrombi usually are composed of tightly coherent masses of platelets, which contain small amounts of fibrin and a few erythrocytes and leukocytes. These thrombi are the classic “white thrombi,” which resemble, in many respects, normal hemostatic plugs. As arterial thrombi enlarge, progressive or intermittent deposition of new layers of platelets and fibrin produces the characteristic lines of Zahn; partial or complete obstruction of blood flow may produce a “tail” of “red thrombus.” The most serious consequences of arterial thrombosis are vascular occlusion with resultant ischemia and infarction of tissue and distal thrombus embolization.
FIGURE 55.1. Pathophysiology of thrombosis. Factors implicated in the pathogenesis of arterial thrombosis (left) and venous thrombosis (right) are depicted. Examples of disorders leading to platelet activation and arterial thrombosis include atherosclerosis, the myeloproliferative disorders, heparin-associated thrombocytopenia/thrombosis syndrome, thrombotic thrombocytopenic purpura, and certain platelet polymorphisms. Examples of disorders leading to venous thrombosis in the category of deficiency of physiologic inhibitors include the inherited disorders, factor V Leiden, proteins C and S deficiencies, and AT deficiency. Many patients with thrombosis may have more than one of the risk factors listed. Estrogen therapy is a risk factor for venous thrombosis; its use is associated with activation of coagulation.
Hypertension, turbulent blood flow at arterial branch points and at sites of focal atherosclerosis, and hyperviscosity may be contributory factors in certain forms of arterial thrombosis. Causes of plasma hyperviscosity that can precipitate thrombosis and exacerbate ischemia include acute myeloid leukemia, myeloproliferative syndromes such as polycythemia rubra vera, cryoglobulinemia, and the plasma cell dyscrasias, including multiple myeloma and Waldenström macroglobulinemia. Immunoglobulin (Ig) paraproteins produced by plasma cell dyscrasias can increase viscosity, and promote red blood cell agglutination.
Venous thrombosis typically develops under conditions of slow blood flow (low shear stress) and is augmented by further retardation and stagnation of flow caused by the developing thrombus itself. Right-sided heart failure, pre-existent venous thrombosis, extrinsic vascular compression by tumor, and chronic venous insufficiency all promote venous stasis, blood pooling, and a concentration of procoagulant factors.8 The anatomic structure of venous valves results in retrograde eddy currents, which produce pockets of stasis even in normal veins.9 The venous arcades in the soleus muscles of the calves may represent another site of physiologic venous stasis. These structures may enlarge and lose vascular tone with aging.10, 11 These facts may partially explain why DVT most commonly occurs in the valve cusps and veins of the pelvis and lower extremities, and why older age is an important risk factor for venous thrombosis.12
Venous thrombi are composed of large amounts of fibrin containing numerous erythrocytes. In these loose, friable masses (the red thrombus), the platelets and leukocytes are enmeshed in random fashion. Venous thrombi resemble blood clots formed in vitro, and they usually produce significant obstruction to blood flow from the outset, but their most serious consequence is embolization. Blood flow obstruction secondary to venous thrombosis itself promotes the further formation of thrombus. Results of studies of clots formed in a thromboviscometer at varying rates of shear suggest that the differences in the structure of venous and arterial thrombi may be mainly the result of the velocity of blood flow. The many and complicated rheologic factors that may be involved in thrombosis have been reviewed.13 Although traditional teaching holds that risk factors for arterial and venous thrombotic disease are distinct, new epidemiologic data suggest that common risk factors exist for both disorders, and that appropriate management of thrombosis patients should address secondary prevention of both arterial and venous thrombosis.14 The pathogenesis of venous and arterial thrombosis has been recently reviewed.15
Vascular Injury
Permanent and transient vascular injuries play major roles in the development of arterial thrombosis. Intraluminal vascular endothelial cell injury, atherosclerotic plaque rupture, hyperhomocysteinemia, arterial outflow obstruction, aneurysm formation, and vessel dissection are among the recognized risk factors for arterial thrombosis.16 Arterial thrombosis usually begins with platelet adhesion to an abnormal vascular endothelial surface or exposed subendothelial constituent such as collagen. The adherent platelets become activated, leading to the release of α– and dense granule contents.15 Platelet-dense granules (dense bodies) release adenosine diphosphate (ADP), adenosine triphosphate (ATP), calcium, and serotonin into the surrounding milieu, resulting in the recruitment and activation of additional platelets.16 This release reaction and platelet synthesis of thromboxane A2 (TxA2) and other agonists induce the aggregation of more platelets and enlargement of the temporary platelet plug. In addition to the recruitment of additional platelets, the original nidus of adherent platelets provides a phospholipid surface rich in phosphatidylserine to support and concentrate the generation of thrombin and fibrin necessary to reinforce and stabilize the platelet plug.
New insights into arterial thrombus formation have been gleaned from experiments using confocal and wide-field microscopy to image real-time thrombus formation in live-mouse cremaster muscle arterioles.17 Thrombosis is precipitated by laser-induced endothelial injury. These experiments demonstrated that the initiation of blood coagulation in vivo involves the initial accumulation of tissue factor (TF) on the upstream and thrombus-vessel wall interface of the developing thrombus. The TF is biologically active and is associated with intrathrombus fibrin generation. TF density is highest at the thrombus-vessel wall interface and is eventually observed throughout the thrombus. Leukocyte rolling is noted approximately 2 minutes after endothelial cell injury and correlates with P-selectin expression on the outer aspect of the thrombus. Minimal TF and fibrin are detected in platelet thrombi formed in mice lacking P-selectin or the P-selectin glycoprotein ligand, PSGL-1.
In venous thrombosis, the luminal surface of the vessel wall is usually histologically normal, and factors extrinsic to the vessel appear to have a major pathophysiologic role. Exceptions to this generalization are direct venous trauma, extrinsic venous compression, and vascular endothelial cell injury resulting from the toxic effect of cancer chemotherapy and excess levels of homocysteine. A limited quantity of activated platelets likely serves as a phospholipid surface to support local thrombin and fibrin formation. A generalized reduction in venous tone may be an important pathophysiologic factor in venous thrombosis in pregnant women and in women taking oral contraceptives.18 vWF-mediated platelet adhesion was shown to be essential for venous thrombosis in an animal model.19 Additional mechanisms may contribute to a prothrombotic state, including up-regulation of stress-response genes, endothelial cell activation, formation of neutrophil extracellular traps, and activation of the intrinsic coagulation pathway.20
Studies indicate that human platelets express TF pre-mRNA, and when activated, splice this message into mature mRNA which is expressed.21 These results suggest that both platelets and the vessel wall may contribute to initiation of thrombosis.
Abnormalities of the Blood: Hypercoagulability
The term hypercoagulable state and its synonym thrombophilia refer to any inherited or acquired abnormality of the hemostatic system that places an individual at increased risk for venous or arterial thrombosis (or both). Blood from patients with active thrombosis or with a hypercoagulable state may clot at an abnormally rapid rate in vitro.22 The concept of hypercoagulability has gained widespread acceptance, and it is generally appreciated that these hemostatic changes are important in the pathogenesis of thrombosis; however, whether testing for these disorders may be helpful in patient management is uncertain.23, 24, 25
Platelet Abnormalities
Although platelets may be incorporated into virtually any thrombus, they appear to be pathogenetically most important in arterial thrombosis. Increased platelet turnover (shortened platelet survival, compensated platelet destruction) occurs in vascular disease and thrombosis, including arterial and venous thrombosis, coronary artery disease, vasculitis, hyperhomocysteinemia, and valvular heart disease.26, 27, 28, 29 Increased platelet turnover can also be seen in patients with risk factors for vascular disease, including those who use tobacco and those with hyperlipidemia.30, 31 Increased platelet turnover and activation can reflect thromboembolic disease and also probably contribute to an exacerbation of thrombotic events.
As a consequence of platelet-vessel wall interactions in patients with vascular disease, platelet adhesion, activation, and aggregation occur, resulting in acquired storage pool disease and the presence in the blood of platelet-granule-derived proteins such as platelet factor-4 (PF4) and β-thromboglobulin.32, 33 Although elevated blood levels of these proteins indicate increased platelet turnover, improper blood collection technique may artifactually result in abnormal levels, and the clinical value of such testing is uncertain.
Vascular endothelium possesses multiple antiplatelet properties that may be important in preventing platelet adhesion, promoting vasodilation, and inhibiting platelet aggregation.34, 35, 36, 37 However, interruption of endothelial cell prostacyclin synthesis by aspirin does not result in a net thrombotic tendency. The potential role of hyperactive platelets in patients with thrombosis, as well as the use of platelet function testing in this setting, is somewhat controversial.
Because of the importance of vWF in mediating platelet adhesion to subendothelium, the role of vWF in human vascular disease has been a topic of investigation. An association between elevated plasma vWF levels and recurrent MI has been demonstrated, and vWF antigen is an independent predictor of coronary artery disease.38
Platelet polymorphisms have been evaluated as risk factors for arterial thrombosis. These include polymorphisms for glycoprotein (gp) IIIa (PlA2), gpIa (807 T allele), and gpIbα.39 The PlA2 polymorphism is linked with coronary artery disease; PlA2-positive platelets exhibit a lower threshold for activation and also have variable aspirin sensitivity.39
Coagulation Abnormalities
Coagulation disorders that can contribute to hypercoagulability can be divided into three risk-factor categories: Transient, inherited, and acquired.40Transient risk factors represent well-defined, temporary clinical circumstances that are associated with increased thrombosis risk both while they are present and for a short period after they have resolved. Examples include surgery, prolonged immobilization, oral contraceptive pill (OCP) use, hormone replacement therapy (HRT), pregnancy, cancer chemotherapy, and heparin-induced thrombocytopenia (HIT). Inherited risk factors represent genetic mutations and polymorphisms that result in deficiency of a natural anticoagulant (e.g., protein C, protein S, or AT), procoagulant factor accumulation (e.g., prothrombin G20210A), or coagulation factor resistance to inactivation by a natural anticoagulant (i.e., factor V G1691A, also known as factor V Leiden). These conditions are characterized by a disruption in the normally highly regulated coagulation mechanism, resulting in greater thrombin generation and an increased risk of clinical thrombosis. Acquired risk factors result either from medical conditions or from nonfamilial hematologic abnormalities that interfere with normal hemostasis or blood rheology. Examples include cancer, inflammatory bowel disease, nephrotic syndrome, vasculitis, antiphospholipid antibodies, myeloproliferative syndromes, paroxysmal nocturnal hemoglobinuria, and hyperviscosity syndromes. These acquired risk factors are distinct from transient risk factors by the fact that they represent alterations in hemostatic homeostasis as a result of disease or, for the most part, nonreversible processes. In contrast, transient risk factors result from either a therapeutic intervention or an adverse reaction to such an intervention. Hyperhomocysteinemia and increased factor VIII functional activity are examples of thrombosis risk factors that can be acquired in nature or have a genetic predisposition.41, 42 Age defies categorization but remains the single most predictable risk factor for thrombosis. The estimated baseline annual age-associated risk of VTE is 1 in 10,000 people <40 years of age, increasing to 1 in 100 people >75 years of age.43, 44, 45
Recent reports have focused on elevated levels of various coagulation factors, particularly fibrinogen and factors V, VII, VIII, X, and XI, that have been documented in patients with thrombosis and prethrombotic disorders, including pregnancy and use of estrogen-containing contraceptives.46 Fibrinogen and factors V and VIII are acute-phase reactants, and their plasma levels may rise in patients with virtually any disorder associated with tissue damage or inflammation, including most thrombotic processes. Whether increased because of a genetic factor or an acute-phase reaction, excess procoagulant factor concentrations may tilt the hemostatic scales in favor of hypercoagulability and may be associated with an increased risk of thrombosis. Epidemiologic studies have identified dose-dependent elevations in plasma levels of vWF, fibrinogen, factor VII, factor IX, and factor XI as independent risk factors for cardiovascular disease.47, 48, 49, 50
Fibrinolytic Abnormalities
Abnormal fibrinolysis has also been linked to vascular disease. Low fibrinolytic activity (measured by dilute clot lysis) was a significant determinant of coronary artery disease in the Northwick Park Heart Study, and elevated PAI-1 activity was associated with major ischemic events in other studies.51, 52 In the European Concerted Action on Thrombosis and Disabilities study, an increased incidence of vascular events was associated with higher baseline PAI-1 antigen concentrations (p = .02) and PAI-1 activity (p = .001).53 Defects in endogenous fibrinolytic capacity have also been found in young patients with severe limb ischemia or unexplained arterial thromboembolism. Deficient t-PA release was found in 45% of such patients, and elevated levels of PAI-1 were found in 59%.54 Excess PAI-1 resulting in impaired endogenous fibrinolysis has also been associated with venous thrombosis in select cases.55
ACTIVATION OF COAGULATION
Mechanisms leading to activation of coagulation in patients with thrombosis have been proposed. TF is thought to be the primary initiator of in vivo coagulation.56, 57 In the absence of TF expression, endothelial cells actively maintain thromboresistance. TF may be expressed in trace amounts during various physiologic processes, such as during normal parturition and after even minimal trauma, including minor head injuries. Immunologic injury of endothelium may lead to the exposure of TF.58 Antibodies formed to exogenous heparin may bind to heparan sulfate on the endothelium, resulting in cell injury, the expression of TF, and the initiation of coagulation.59 In a similar manner, endothelial cells may be induced to express TF by interleukin-1, homocysteine, tumor necrosis factor, and endotoxin.60, 61, 62, 63 TF antigen expression by endothelium has been demonstrated in pathologic primate and human tissues.64, 65, 66 Activation of coagulation may also occur on monocytes, and platelets can be activated to mediate factor-Xa-catalyzed prothrombin conversion to thrombin.67, 68 Deficient hepatic clearance of activated coagulation factors may represent an important thrombogenic factor in premature infants and in patients with liver disease, especially after administration of prothrombin complex concentrates that contain trace amounts of activated vitamin-K-dependent coagulation factors.
Various other mechanisms may be responsible for low-grade in vivo coagulation under certain conditions. Certain tumor cells can promote thrombin generation directly, by producing TF, expressing the coagulation factor X activator known as cancer procoagulant, or displaying surface sialic acid residues that can support nonenzymatic factor X activation.69, 70 Tumor cells can also promote thrombin generation indirectly, by eliciting TF expression by monocytes and endothelial cells. Tumor-associated coagulation cascade activation can result in the generation of thrombin that can promote angiogenesis by signaling through protease-activated receptors PAR-1, PAR-3, and PAR-4. In addition to its normal role in coagulation, evidence is mounting that TF plays a direct role in growth, invasion, dissemination, and angiogenesis of tumors.71, 72 Clotting-independent signaling through the TF cytoplasmic tail can lead to vascular endothelial growth factor (VEGF) up-regulation. Because VEGF can promote TF up-regulation, the potential for a self-perpetuating proangiogenic amplification loop exists. The TF:factor VIIa complex can also activate PAR-2 directly in a manner that results in gene expression related to angiogenesis and cell migration.72 Selected tumors may mediate an accentuation of platelet activation and accumulation, whereas other tumor cells may express surface phospholipid species such as phosphatidylserine, which can support prothrombin and factor X activation.73
A laboratory picture similar to that described in the hypercoagulable state may also be seen in patients with nonovert (compensated or chronic) disseminated intravascular coagulation (DIC).74 In such patients, the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) may be shortened because of the presence of traces of thrombin or other activated coagulation factors. These activated factors affect one-stage, but not two-stage, clot endpoint coagulation assays. High levels of factor V and factor VIII activity may reflect the presence of thrombin-activated forms of these factors in the circulation. Pregnancy, which can be regarded as a hypercoagulable state, has also been described as a physiologic form of nonovert DIC by some clinicians.75 Pregnancy increases the risk of venous thromboembolism (VTE) approximately fivefold among the general population, and female patients who have experienced prior venous thrombosis have a threefold risk of venous thrombosis during pregnancy.76 The use of estrogen-containing oral contraceptives and estrogen HRT is clearly associated with an increased risk of VTE.77, 78, 79 A prospective study of these patients indicates that estrogens induce activation of coagulation as well as reduction in levels of natural anticoagulants such as AT and protein S.80 However, many patients who experience VTE while taking oral contraceptives have a common inherited thrombotic disorder, APC resistance (APC-R), or prothrombin G20210A.81 Another report indicated that oral contraceptives induced “acquired APC-R” in women.82 The relative risk of VTE in postmenopausal women using estrogen replacement is 2.14, and the risk is highest during the first year of use.83 The thrombotic risk imparted by pharmacologic estrogens appears to dissipate following medication discontinuation.84 One study found that transdermal estrogen products were safer than oral estrogens in women receiving HRT.85 One literature review of hormonal therapy and venous thrombosis concluded that all oral estrogen-containing products used for contraception or HRT, as well as therapeutic doses of progesterone-only products increased thrombosis risk.79 The risk of venous thrombosis is not clearly increased for the levonorgestrel-containing intrauterine device, transdermal estrogen, and tibolone.79 Regarding the risk of thrombotic stroke and MI, a very large Danish study found that use of ethinyl estradiol increased risk by 0.9 to 2.3, depending on drug concentration in the contraceptive pill.86Table 55.1 summarizes acquired etiologies that predispose to thrombosis.
INHERITED THROMBOTIC DISORDERS
Several monographs and reviews have covered the topic of inherited thrombotic disorders in detail.40, 46, 86, 87, 88Table 55.2 summarizes the prevalence of selected inherited and acquired hypercoagulable states in different populations, whereas Table 55.3 summarizes the clinical presentations of venous thrombosis that may be suggestive of particular hypercoagulable states. Many patients who experience thrombosis are found to have a combination of defects, for example, APC-R plus use of oral contraceptive agents,81 or a combination of inherited defects.40 Although people with inherited hypercoagulable states are at a greater risk for developing a thrombotic event than those without such disorders, not all people with a well-defined hypercoagulable state develop an overt thrombosis, and not all people with thrombosis have an identifiable hypercoagulable state. Testing for an inherited hypercoagulable state is likely to uncover an abnormality in >60% of patients presenting with idiopathic (i.e., spontaneous or unprovoked) venous thrombosis.89 Although the remaining 30% to 40% have unremarkable test results, this does not imply a true absence of a hypercoagulable state. Some of these individuals may have an acquired condition such as cancer or antiphospholipid antibodies, whereas others may have a disorder or genetic defect that has not yet been discovered or characterized.
TABLE 55.1 ACQUIRED DISORDERS PREDISPOSING TO THROMBOSIS
Adenocarcinoma (particularly of the gastrointestinal tract)
Neonatal purpura fulminans
Homozygous protein C or protein S deficiency
PT, prothrombin.
Antithrombin Deficiency
AT, formally known as AT-III, deficiency was first described by Egeberg in 1965.90 AT, a serine protease inhibitor, regulates coagulation by inactivating thrombin and other procoagulant enzymes, including factors Xa, IXa, XIa, and XIIa. As are most inherited thrombotic disorders, AT deficiency is inherited as an autosomal dominant disorder.91, 92 A blood bank survey reported that 1 in 600 people has AT deficiency.93 The interactions of unfractionated and low-molecular-weight heparin (LMWH) with AT are discussed in the section “Heparin.”
Pathophysiology and Genetics
Patients with AT deficiency may have either type I (quantitative deficiency) or type II (qualitative abnormality) disease. Type I AT-deficient patients usually have concordant reductions in AT measured by both immunologic and functional assays. Type II patients have reduced functional AT activity associated with normal amounts of AT protein. Plasma concentrations of the thrombin activation peptide, prothrombin fragment 1+2, are elevated in patients with AT deficiency, indicating persistent activation of coagulation as a result of deficient neutralization of factor Xa in these patients.94
The genetic basis for type I AT deficiency is either deletion of a gene segment or the occurrence of point mutations or deletions, resulting in a nonsense mutation and an incomplete protein. The genetic basis for type II AT deficiency is the occurrence of point mutations that do not impair synthesis of the protein, but that result in a dysfunctional protease inhibitor. A summary of AT mutations (>100) has been reported.95 A database of AT mutations is located at: http://www1.imperial.ac.uk/departmentofmedicine/divisions/experimentalmedicine/haematology/coag/AT/. In general, mutant AT molecules exhibit deficient heparin binding, deficient protease inhibition, or both.
Clinical Aspects
AT deficiency is manifested primarily by recurrent VTE.91, 92 Almost every vein site has been reported to be involved with thrombosis in AT-deficient patients, including unusual sites such as mesenteric vessels.96 Thrombosis may occur in the absence of precipitating factors or may result from events such as pregnancy, estrogen use, trauma, or surgery. AT deficiency can result in heparin resistance manifesting as a normal to minimally increased aPTT in patients receiving large doses of heparin.97 However, most patients with AT deficiency do not exhibit heparin resistance, and most cases of heparin resistance are not due to AT deficiency. In family studies, venous thrombosis occurred in 85% of AT-deficient relatives before 55 years of age.97 The estimated increased lifetime relative risk of venous thrombosis has been reported to be up to 40-fold.98, 99 An annual absolute risk of venous thrombosis of 27.5% has been reported in female carriers of AT deficiency who also use OCPs.100 Early studies reported pregnancy-related venous thrombosis rates as high as 70% for AT-deficient women.101, 102 Most AT-deficient patients are heterozygotes and possess ˜50% of normal activity levels. Although homozygotes have been reported in consanguineous kindreds, in general, homozygous type I AT deficiency is believed to be fatal in utero.103
Laboratory Diagnosis
Results of the PT and aPTT are normal in AT-deficient patients. A variety of commercial assays is available to measure AT levels, including functional and immunologic assays. Functional assays are preferable because they detect both type I and type II patients. One report found that 2% of patients with AT deficiency had type II disease.104 Functional assays for AT measure heparin cofactor activity using a chromogenic substrate method to quantitate thrombin or factor Xa neutralization.105 A potential disadvantage of functional AT assays is that plasma AT levels may be overestimated because of the presence of heparin cofactor II (HCII) in the sample. The use of bovine thrombin and a low heparin concentration (3 U/ml) in the assay system can minimize this problem.106
Immunologic assays for AT use Laurell rocket immunoelectrophoresis, microlatex-particle immunoassay, radial immunodiffusion, or enzyme-linked immunosorbent assay (ELISA) methods.107 Ideally, patients should be evaluated for AT deficiency at a time when they are not receiving therapeutic heparin or oral anticoagulants, because heparin depresses AT levels, whereas long-term warfarin therapy increases plasma AT levels in some patients.92, 108 Laboratory tests for AT deficiency have been summarized.109
Treatment
Some AT-deficient patients may experience heparin resistance, requiring the administration of AT by using virally inactivated, plasma-derived, or transgenic AT concentrates, or fresh-frozen plasma. Supplemental AT may make it easier to achieve therapeutic anticoagulation with heparin in these patients.110 Patients with recurrent thrombosis should receive long-term warfarin therapy at a dosage to maintain an international normalized ratio (INR) value of 2.0 to 3.0 (discussed in the section “Warfarin”). Patients with a single thrombotic event should receive at least 3 to 6 months of warfarin therapy and, because of an increased recurrence rate, should be considered for long-term therapy beyond 6 months. Patients with massive venous thrombosis or PE may be candidates for thrombolytic therapy. AT-deficient patients who become pregnant or who will undergo general surgery should be considered for anticoagulant prophylaxis, including AT concentrate administration.110, 111 Most asymptomatic patients should not be treated. The avoidance of estrogens should be considered in AT-deficient patients, who are not therapeutically anticoagulated.
Acquired Antithrombin Deficiency
AT deficiency is also associated with numerous disorders, including DIC, liver disease, nephrotic syndrome, and preeclampsia, and is seen in patients taking oral contraceptive agents and during pregnancy.112, 113, 114, 115 Although logic suggests that correction of AT deficiency might be clinically useful in disorders such as DIC or liver disease, there are no conclusive data supporting AT replacement therapy outside the setting of prophylaxis of high-risk patients, such as pregnant, AT-deficient patients.116
Heparin Cofactor II Deficiency
HCII is another heparin-dependent thrombin inhibitor, differing from AT in that a glycosaminoglycan other than heparin, dermatan sulfate, catalyzes this inhibitor of coagulation.117 HCII deficiency is inherited as an autosomal dominant trait. Although there have been anecdotal studies of thrombosis associated with HCII-deficient kindreds, a larger study has concluded that HCII deficiency by itself may not be an inherited risk factor for thrombosis.118, 119, 120 Commercial assays to measure HCII levels are available, but the clinical utility of such assays in routine evaluation of patients for inherited thrombosis is uncertain.
Protein C Deficiency
Protein C is a vitamin-K-dependent plasma protein that, when activated by the thrombin-thrombomodulin complex to APC, inactivates factors Va and VIIIa to inhibit coagulation.121 APC also possesses profibrinolytic activity that results from neutralization of PAI-3 activity.122 Inherited deficiency of protein C and its association with thrombosis were first described by Griffin and coworkers in 1981.123 Protein C deficiency was believed to be inherited in an autosomal dominant pattern with incomplete penetrance. More recent studies have suggested that protein C deficiency may be an autosomal recessive disorder and that coinheritance of another defect (particularly factor V Leiden) results in a high degree of penetrance that appears as the dominant inheritance in double-heterozygous carriers.124, 125
Pathophysiology and Genetics
As with AT deficiency, patients with protein C deficiency may have type I (quantitative deficiency) or type II (qualitative abnormality) disease. Most patients are heterozygotes with ˜50% of normal protein C levels. A hypercoagulable state can be demonstrated in nonanticoagulated protein-C-deficient patients using activation peptide (fragment 1+2) assays.126
In type I protein C deficiency, more than half of the gene mutations identified are missense mutations. Point mutations affecting protein function appear to be common in patients with type II protein C deficiency. At least 195 different gene abnormalities have been associated with both types of protein C deficiency.127 Despite the clear association of protein C deficiency with thrombosis in large epidemiologic studies, there are also definitive data indicating that many protein-C-deficient patients are asymptomatic.128 For example, one report found that heterozygous protein C deficiency occurred in approximately 1:250 subjects, whereas a large Scottish study estimated the prevalence to be approximately 1:500.124, 129 No patient in either study had a history of symptomatic venous thrombosis. These findings indicate that additional risk factors—acquired, genetic, or both—are necessary to provoke thrombosis in heterozygous protein-C-deficient patients. Indeed, in one study, up to 20% of symptomatic protein-C-deficient patients also had APC-R, supporting the concept that many patients with recurrent thrombosis have more than one risk factor.125
Clinical Aspects
Three clinical syndromes are associated with protein C deficiency: VTE in heterozygous adults, neonatal purpura fulminans in homozygous newborns, and warfarin-induced skin necrosis in certain heterozygous adults. The predominant clinical symptom of protein-C-deficient patients is recurrent VTE, although arterial thrombotic events, including stroke, have been reported.130, 131 As mentioned above, many patients are found to have risk factors other than inherited protein C deficiency on investigation, such as APC-R, use of oral contraceptive agents, and pregnancy. Protein C deficiency has also been linked to fetal loss.132
Neonatal purpura fulminans is seen in homozygous newborns of heterozygous parents. These children develop DIC at birth, associated with extensive venous or arterial thrombosis (or both) and very low levels of protein C (<5% of normal).133 Warfarin-induced skin necrosis is an unusual syndrome seen in certain patients with heterozygote protein C deficiency.134 Most patients who develop this syndrome have received large doses of warfarin in the absence of concomitant overlapping therapeutic parenteral anticoagulation. The basis for this syndrome is that warfarin therapy, especially in large loading doses, reduces protein C levels more rapidly than the vitamin-K-dependent procoagulant factors, leading to exacerbation of the basal hypercoagulable state and thrombosis.
In family studies, venous thrombosis occurred in 50% of protein-C-deficient relatives of affected probands before 40 years of age.135 The estimated increased lifetime relative risk of venous thrombosis has been reported to be up to 31-fold.98 As stated above, other studies did not detect an increased thrombosis risk in carriers of protein C deficiency. Differences in risk between family- and population-based studies can be in part explained by greater difficulty in obtaining reliable population-based estimates because of the overall low prevalence of this and other natural anticoagulant deficiencies. In studies of unselected patients with venous thrombosis, the odds ratio (OR) of having protein C deficiency is increased six- to ninefold compared to controls.40, 135, 136, 137 An annual absolute risk of venous thrombosis of 4.3% has been reported in women carriers of a natural anticoagulant deficiency such as protein C deficiency who also use OCPs.100, 138
Laboratory Diagnosis
Patients with heterozygous protein C deficiency have normal PT and aPTT values, whereas patients with homozygous protein C deficiency have abnormal coagulation tests consistent with DIC. Because both type I and type II disorders may occur, functional assays are suggested to optimize identification of affected patients. Some investigators prefer the clot endpoint-based assay, because it measures complete function of the protein C molecule, including those patients with abnormal protein C molecules that possess normal activity by a chromogenic substrate assay. However, therapeutic heparin levels affect the clot-based assay. A College of American Pathologists’ consensus conference on thrombophilia recommends use of the chromogenic substrate assay.139 One report indicated that measurement of the ratio of protein C to protein S antigen was useful in identifying carriers of protein C deficiency.140
Protein C levels can also be measured by immunologic methods, including Laurell rocket immunoelectrophoresis and ELISA. However, immunologic assays may not detect type II patients and may overestimate protein C levels in warfarin-treated patients. Immunologic assays may be more useful in evaluating patients with homozygous deficiency and DIC.
Age-related changes occur with protein C levels.141 Consequently, it is important to consider this when testing younger patients (<30 years); otherwise, normal subjects may be misclassified as protein C deficient.
A common problem faced by laboratories is measuring protein C levels in patients taking oral anticoagulants. Many clinicians forget that protein C is a vitamin-K-dependent molecule, and that otherwise hemostatically normal people taking warfarin may have low protein C levels. Griffin and coworkers suggested that protein C data be normalized against the level of another vitamin-K-dependent protein to distinguish inherited protein-C-deficient patients from normal subjects taking warfarin.123 One author reported that using a ratio between protein C and prothrombin optimally categorized patients.142 However, for this method to be useful, patients must be stably anticoagulated for at least 2 weeks, and the laboratory must obtain plasma samples and reference ranges from patients taking warfarin for reasons other than recurrent thrombosis. The consensus conference did not recommend assaying protein C levels in patients on oral anticoagulants.139 Laboratory tests for protein C deficiency have been reviewed.143
Treatment
Many patients with protein C deficiency are asymptomatic, especially those identified in screening studies. Asymptomatic patients should not be treated but should be considered for prophylaxis when they experience high-risk procedures, such as surgery. Symptomatic protein-C-deficient patients should be anticoagulated with heparin and then considered for long-term secondary prophylaxis with warfarin at an INR of 2.0 to 3.0. Those patients with a single thrombotic event should receive at least 3 to 6 months of warfarin. Patients with more than one thrombotic event, patients with a single life-threatening thromboembolic event, and those with a significant family history of thrombosis should be considered for long-term anticoagulation. Patients with massive thrombosis or PE may be candidates for thrombolytic therapy.
Infants with neonatal purpura fulminans should be treated with protein C replacement therapy. Protocols using a purified protein C concentrate or prothrombin complex concentrates have been described.144, 145 Use of the purified concentrate normalizes activation of coagulation in these homozygous patients.146
Acquired Protein C Deficiency
Because protein C is a vitamin-K-dependent protein, any disorder associated with vitamin K deficiency may result in protein C deficiency, including warfarin use, liver disease, and malnutrition.147 Protein C levels are also reduced in DIC, presumably reflecting thrombin activation of the zymogen and consumption of APC.148 Protein C levels may also be reduced in renal disease, especially the nephrotic syndrome.149
Protein S Deficiency
Protein S is a vitamin-K-dependent plasma protein that facilitates the anticoagulant activity of APC. Protein S deficiency in association with inherited thrombotic disease was first described by two groups in 1984.150, 151 As with AT and protein C deficiencies, protein S deficiency is inherited as an autosomal dominant trait. Many patients with a previous diagnosis of protein S deficiency actually have APC-R.152, 153 This diagnostic error results from interference in the functional protein S assay of patients with the inherited disorder, APC-R, and misclassification of patients. These patients have a “pseudo” protein S deficiency, often display a type II deficiency pattern, and have protein S activity levels that correlate with the APC ratio. Based on these reports and the high incidence of APC-R in the general population, the true importance of protein S deficiency in inherited thrombosis is uncertain.152, 153, 154
Pathophysiology and Genetics
As described for AT and protein C deficiencies, patients with protein S deficiency may have quantitative or qualitative disorders. Under normal circumstances, protein S exists in plasma in two forms: Bound to C4b-binding protein (60% of total protein S) and free (40% of total). Because only free protein S has APC co-factor activity, a revised classification system has been proposed for protein S deficiency.155 Type I protein S deficiency is a quantitative disorder in which protein S functional activity, total antigen, and free antigen levels are equally reduced to ˜50% of normal. Type IIa protein S deficiency is a deficiency of free protein S with preserved normal levels of total protein S. In type IIb protein S deficiency, the levels of both total and free protein S antigen are normal. Apparent type IIb protein S deficiency has been described in patients with APC-R.154 An acquired type IIa protein S deficiency may result from excess levels of C4b-binding protein or the presence of free protein S inhibitory and clearing autoantibodies.156 Almost 200 different mutations have been identified in the gene that codes for protein S.95, 157 A full listing of protein S gene mutations and polymorphisms can be found at www.ISTH.org.
As with protein C deficiency, many patients with protein S deficiency and thrombosis have additional risk factors. In one study, among patients with protein S deficiency, ˜40% also had APC-R; of family members with thrombosis, 72% had both defects, whereas <20% of patients with single defects experienced thrombosis.158
Clinical Aspects
Like AT and protein-C-deficient patients, most patients with protein S deficiency and thrombosis have experienced VTE.150,151 However, unlike most other inherited thrombotic disorders, up to 25% of patients with protein S deficiency may experience arterial thrombosis, including stroke.131, 159 As mentioned previously, many patients with protein S deficiency and thrombosis have other risk factors, including APC-R, estrogen use, or pregnancy.158 Neonatal purpura fulminans, fetal loss, and warfarin-induced skin necrosis have also been associated with protein S deficiency.132, 160 In family studies, venous thrombosis occurred in 100% of protein-S-deficient relatives of affected probands by 70 years of age.161 The estimated lifetime increased relative risk of thrombosis has been reported to be as high as 36-fold for protein S deficiency.98
Laboratory Diagnosis
Patients with heterozygous protein S deficiency have normal PT and aPTT values. The laboratory diagnosis of protein S deficiency is complicated by four factors: The levels of C4b-binding protein, the coexistence of APC-R in certain patients, elevated factor VIII activity levels, and warfarin therapy. C4b-binding protein is an acute-phase-reactant protein-S-binding protein, often elevated in thromboembolism, resulting in reduced free protein S levels from the patient’s true baseline. Consequently, measurement of protein S levels in patients with acute thrombosis may yield misleading results. Similarly, false-positive results may be seen when functional protein S assays are performed on patients who have APC-R or factor VIII activity levels ≥250%.155 Patients should not be assumed to have protein S deficiency (diagnosed by functional assay) until APC-R has been excluded. Lastly, because protein S is a vitamin-K-dependent protein, warfarin therapy and vitamin K deficiency pose the same difficulty as described for patients evaluated for protein C deficiency. Protein S levels may also be decreased in the setting of pregnancy but are not necessarily associated with thrombotic events.
Both functional and immunologic assays are commercially available to quantitate plasma protein S levels. Functional assays may be PT- or aPTT-based, measuring inhibition of factor Va by APC.162 These assays have the advantage of measuring free protein S activity. Immunologic assays to measure either total (free plus C4b-binding-protein bound) or free protein S levels are available.163, 164 Immunologic assays may be useful in evaluating patients who have co-existing APC-R. Total protein S levels are measured by Laurell rocket immunoelectrophoresis or ELISA. In the interpretation of immunologic assays, one should consider the report that the mean plasma level of protein S antigen in males is higher than it is in females.164
Treatment
Asymptomatic patients should not be treated but should be considered for prophylaxis when they experience high-risk procedures such as surgery. Symptomatic protein-S-deficient patients should be anticoagulated as described for protein-C-deficient patients.
Protein Z Deficiency
Protein Z is a vitamin-K-dependent plasma protein that serves as a co-factor for activated factor X inhibition by the protein-Z-dependent protease inhibitor.165 Reduced circulating levels of protein Z have been implicated in thrombosis.166, 167 Protein Z deficiency has also been linked to early and late fetal demise and intrauterine growth restriction.167 Protein Z deficiency has also been found in the setting of acute coronary syndromes, whereas a deficiency state and the presence of a series of protein Z variants that modulate protein Z plasma levels have been associated with stroke.167, 168, 169, 170 Further data are required before routine protein Z testing can be recommended.
Activated Protein C Resistance (Factor V Leiden)
Before 1993, most patients with idiopathic venous thrombosis evaluated for inherited thrombosis were not given a diagnosis, because AT, protein C, and protein S deficiencies together were found in <20% of patients. In 1993, Dahlback and colleagues in Sweden postulated that certain patients with recurrent thrombosis might have additional abnormalities of the protein C pathway, resulting in a hypercoagulable state.171 They found that addition of APC to plasma obtained from patients with recurrent thrombosis did not prolong the aPTT to the same degree as that seen when APC was added to normal plasma.171 These patients did not have any previously recognized inherited thrombotic disorder. The term APC resistance was used for these patients. Other investigators then used the aPTT-based APC screening test to examine other populations for this phenotype. APC-R was found in 20% to 60% of patients with recurrent thrombosis.172, 173, 174 Like other inherited thrombotic disorders, APC-R was inherited in an autosomal dominant manner.
Pathophysiology and Genetics
APC inactivates factor Va in an orderly and sequential series of cleavages, first at Arg506, and then at Arg306 and Arg679.175, 176, 177 Although the affected factor V cleavage site in APC-R is not directly responsible for complete inactivation of factor Va, APC cleavage at this site is necessary for subsequent proteolytic events. This “partial resistance” is explained by the fact that cleavage of factor Va by APC at Arg306 continues to occur, albeit at a slower rate.178 In fact, factor V Arg506Gln (factor V Leiden) is inactivated 10 times more slowly than normal factor Va. This provides a pathophysiologic explanation for why factor V Leiden, although common, is a relatively weak risk factor for VTE. Because factor Va functions as a cofactor in the conversion of prothrombin to thrombin, the mutation results in greater amounts of factor Va being available for coagulation reactions, “shifting” the hemostatic balance toward greater thrombin generation.95
APC-R due to factor V Leiden is the most common inherited predisposition to hypercoagulability in Caucasian populations of northern European background.95 Factor V Leiden follows a geographic and an ethnic distribution: The mutation occurs most frequently in northern and western Europe (the highest prevalences, 10% to 15%, have been reported in Cyprus, Sweden, and Turkey) but is rare in the Asian and African continents as well as in ethnic groups of Asian descent, such as Inuit Eskimos, Amerindians, Australian Aborigines, and Polynesians.179 In the United States, factor V Leiden is most commonly seen in Caucasians (6.0%), with lower prevalences in Hispanics (2.2%), African and Native Americans (1.2%), and Asian Americans (0.45%).180
Factor V Leiden accounts for 92% of cases of APC-R, with the remaining 8% of cases resulting from pregnancy, oral contraceptive use, cancer, selected antiphospholipid antibodies, plasma glucosylceramide deficiency,181, 182, 183 and other factor V point mutations. Therefore, the terms factor V Leiden and APC-R should not be considered synonymous; in fact, APC-R is an independent risk factor for VTE even in the absence of factor V Leiden.184 It is estimated that the mutation arose in a single Caucasian ancestor some 21,000 to 34,000 years ago, well after the evolutionary separation of non-Africans from Africans (˜100,000 years ago) and of Caucasoid (white Caucasians) from Mongoloid (Asians) subpopulations (˜60,000 years ago).185
Clinical Aspects
Heterozygous carriers of factor V Leiden have a 2- to 10-fold increased lifetime relative risk of developing VTE.98, 138, 172, 186, 187, 188, 189, 190 This risk is further increased in combination with pregnancy (9-fold), OCP use (36-fold), and HRT (13- to 16-fold).191, 192, 193, 194 VTE is the most common clinical symptom of APC-R in patients who experience thrombosis. In general, there is a notable lack of association of APC-R with arterial thrombosis (exceptions discussed below).188 Another clinical association with APC-R is recurrent miscarriage, with one study reporting that 20% of patients with second-trimester pregnancy loss have APC-R.132, 195 Factor V Leiden does not appear to be a cause of recurrent pregnancy loss occurring late in the first trimester.195, 196 Neonatal purpura fulminans has been reported in a patient with factor V Leiden who did not have protein C or S deficiency.197 APC-R is also a common inherited risk factor for cerebral venous thrombosis.198 The factor V Leiden mutation has also been identified in children who experience thrombosis.199 However, this mutation does not appear to play a major role in the hypercoagulability of cancer.200 An intriguing report has demonstrated that the factor V Leiden mutation is a risk factor for MI in young women; the combination of factor V Leiden with smoking increased the risk of MI more than 30-fold.201 Factor V Leiden has also been associated with MI in individuals with coronary artery thrombosis in the absence of evidence of underlying fixed atherosclerotic lesions.202
There are conflicting data on the role of factor V Leiden heterozygosity as an independent risk factor for VTE recurrence.203, 204 Two studies (“positive studies”) have found an increased risk of VTE recurrence compared to control subjects, with relative risk of 4.1 and 2.4, respectively. Six other studies (“negative studies”) have found no such association.205, 206, 207, 208, 209, 210 The positive studies by Ridker et al. and Simioni et al. were prospective but included a small number of patients with factor V Leiden heterozygosity (14 and 41, respectively).203, 204 The Physicians’ Health Study (by Ridker et al.) included only men.203 Among the negative studies, four were prospective and two were retrospective.205, 206, 207, 208, 209, 210 Three of the prospective studies and the large retrospective cohort study by De Stefano et al. each included 80 to 112 patients with factor V Leiden heterozygosity.205, 206, 208, 210 A recent re-evaluation of the study by Simioni et al. did confirm the original study findings of increased relative risk of recurrent VTE in heterozygous carriers of factor V Leiden.211 The PREVENT trial later demonstrated equivalent rates of recurrent VTE in patients with and without factor V Leiden.212
Homozygous carriers of the factor V Leiden mutation are estimated to have an 80-fold increased lifetime relative risk of VTE.188 A more recent estimate, derived from a pooled analysis of a larger population, has confirmed an increased risk of VTE but of lower magnitude (10-fold).191 The discrepancy is likely due to the very low prevalence of factor V Leiden homozygosity found in the healthy controls from the general population. Most homozygous carriers present with VTE before 40 years of age, but some can live thrombosis-free until the sixth or seventh decade of life or even remain asymptomatic for life.213, 214 The majority of VTE is situational, and women appear more likely to develop VTE than men, suggesting an important role of OCP use and pregnancy in triggering thrombosis.213, 214, 215 Based on data from the first prospective Duration of Anticoagulation trial, the risk of VTE recurrence is significantly increased in homozygous factor V Leiden carriers (36.4% at 48 months) when compared to heterozygous carriers (16.1%) and controls (12.4%).206
Laboratory Diagnosis
The intense interest in this common inherited thrombotic disorder has focused substantial attention on laboratory methods for its diagnosis. Laboratory aspects of its discovery have been reviewed by Dahlback.216 The disorder can be evaluated by coagulation assays that have as their basis inhibition of factor Va by APC and prolongation of the clotting time. Typically, APC is added to patient plasma, and a clotting assay is performed (usually the PTT), with results expressed as a ratio:
Reference ranges are established for normal patients with and without addition of APC. Affected patients with mutant factor V have clotting times prolonged to a lesser extent (lower ratio) than normals. Alternatively, a DNA test can be done to look specifically for the Arg506Gln mutation; this highly conserved point mutation is present in most patients with APC-R.
With properly collected plasma samples, the APC-R clotting test can correctly classify nearly 100% of patients, when normalized to a control plasma pool.217 Predilution of the patient sample with factor-V-deficient plasma has improved the performance characteristics of most currently available commercial assays.218 Samples from patients with baseline abnormal coagulation studies (e.g., anticoagulant therapy, lupus anticoagulants, liver disease) yield uninterpretable results. A TF-dependent factor V assay has been described that is useful in patients taking oral anticoagulants or with the lupus anticoagulant.219 The polymerase chain reaction test for factor V Leiden uses the restriction enzyme MnlI to digest a 267-base pair amplified fragment of patient DNA.176 Consensus recommendations on methodologies to assay for factor V Leiden and APC-R have been presented.220, 221
Treatment
Therapy of VTE in patients with APC-R is similar to that described for patients without an identified hypercoagulable state. Long-term secondary prophylaxis is not necessary for heterozygotes unless they experience more than one thrombotic event or experience life-threatening thromboembolism. Asymptomatic patients with APC-R should not be treated, but female patients with this disorder should be informed about the additional thrombotic risk associated with oral contraceptive use, pregnancy, and HRT.82, 222 Prophylaxis for high-risk situations, such as surgery, should be given. Homozygotes who experience VTE should be considered for long-term anticoagulation.
Prothrombin Mutations
The prothrombin G20210A mutation is the second most common inherited predisposition to hypercoagulability. Heterozygous prothrombin G20210A has been found in 18% of probands of thrombophilic families, 6% of unselected patients with deep vein thrombosis (DVT), and 2% of normal Caucasian individuals.40, 223
More recently, a novel single-point mutation of the prothrombin gene at position 20209 has been reported in four unrelated patients, two of whom had a history of VTE and one of whom had a history of stroke.224 Although the clinical significance of the prothrombin C20209T mutation is unknown, it may be underrecognized because it is not detected by the polymerase chain reaction/digestion assay commonly used for prothrombin gene mutation testing. Interestingly, all four reported individuals with prothrombin C20209T were African-Americans.
Pathophysiology and Genetics
Prothrombin G20210A is a single-point mutation (G-to-A substitution at nucleotide 20210) in the 3′ untranslated region of the prothrombin gene.223 This autosomal dominant mutation results in elevated concentrations of plasma prothrombin.223 In fact, the VTE risk increases as the plasma prothrombin level increases, with levels >115 IU/dl leading to a 2.1-fold increased relative risk of VTE.223 The G20210A mutation leads to a “gain of function” of the prothrombin gene, perhaps by resulting in an altered polyadenylation pattern in mutant prothrombin mRNA.225 An in vitro study of thrombin generation found that increasing prothrombin levels to 150% of normal resulted in enhanced thrombin activity.226
The mutation appears to follow a geographic and ethnic distribution, with the highest prevalence occurring, unlike factor V Leiden, in Caucasians from southern Europe (3%).227 This prevalence is nearly twice that observed in northern Europe (1.7%).227 Similar to factor V Leiden, the prothrombin G20210A mutation is also found in the Middle East and Indian regions, but it is virtually absent in individuals of Asian and African backgrounds.227 These distributions provide support to the estimate that both mutations (factor V Leiden and prothrombin G20210A) originated relatively recently in the European founding population, after the evolutionary divergences of subpopulations. An evaluation of patients in northeast Ohio who underwent hypercoagulability evaluations revealed an equivalent prevalence of prothrombin gene mutations in whites and blacks.224
Clinical Aspects
Heterozygous prothrombin G20210A is associated with a two- to sixfold increased lifetime relative risk of VTE.40, 214, 223, 228 The risk appears to be further increased in combination with pregnancy (15-fold) and OCP use (16-fold).191, 229 The relative risk of cerebral vein thrombosis is increased 10-fold in women with this mutation who are not on OCPs, as opposed to 150-fold in OCP users.230 Homozygosity for prothrombin G20210A has an estimated population prevalence of 0.014%, and homozygous carriers appear to have greater predisposition to develop early (before 40 years of age) idiopathic recurrent VTE than heterozygotes.223, 231, 232
The role of prothrombin G20210A as a risk factor for VTE recurrence is less controversial than it is for factor V Leiden, but data are also somewhat conflicting. Simioni et al., re-evaluating a prospective study from 1997, retrospectively determined patients’ prothrombin G20210A mutation status and found that the hazard ratio of VTE recurrence was 2.4.211 However, three prospective studies, which each included 28 to 52 patients, found no increased risk of recurrent VTE in heterozygous prothrombin G20210A carriers.206, 233, 234
A fourfold increased risk of MI has been demonstrated, particularly in young women carriers of this mutation. A large case-control study of >14,000 men, though, revealed no increased risk of stroke or MI associated with this abnormal prothrombin gene. The reasons for this apparent male/female disparity are unknown.201, 235
Laboratory Diagnosis
The prothrombin G20210A and C20209T mutations can only be reliably and routinely identified using molecular biologic techniques. Measurements of functional prothrombin activity do not sufficiently differentiate between carriers and noncarriers of this gene mutation. Testing can be performed accurately despite concomitant treatment with any form of anticoagulation.
Treatment
Treatment paradigms for patients with prothrombin G20210A heterozygosity parallel those for patients with heterozygous factor V Leiden. Patients with concomitant prothrombin G20210A heterozygosity and factor V Leiden heterozygosity should be considered for long-term anticoagulation following a first thrombotic event.
Hyperhomocysteinemia
Homocysteine is a sulfhydryl amino acid formed during the conversion of methionine to cysteine. Hyperhomocysteinemia results when homocysteine metabolism is abnormal. Hyperhomocysteinemia has been identified as an independent risk factor for stroke, MI, peripheral arterial disease, and venous thrombotic disease.236, 237, 238 Even mild to moderate hyperhomocysteinemia is a significant risk factor for vascular disease. However, lowering homocysteine levels with vitamin therapy has not resulted in improved outcomes in vascular disease and thrombosis.239, 240
Pathophysiology and Genetics
The amino acid homocysteine is normally metabolized via the transsulfuration pathway by the enzyme cystathionine-β-synthase (CBS), which requires vitamin B6 as co-factor, and via the remethylation pathway by the enzymes methylenetetrahydrofolate reductase (MTHFR), which is folate dependent, and methionine synthase, which requires vitamin B12 as co-factor.241, 242 Inherited severe hyperhomocysteinemia (plasma level >100 µmol/L), as seen in classic homocystinuria, may result from homozygous MTHFR and CBS deficiencies and, more rarely, from inherited errors of cobalamin metabolism.241, 242 Inherited mild to moderate hyperhomocysteinemia (plasma level >15 to 100 µmol/L) may result from heterozygous MTHFR and CBS deficiencies, but most commonly results from the thermolabile variant of MTHFR (tlMTHFR) that is encoded by the C677T gene polymorphism.241, 242
Acquired hyperhomocysteinemia may be caused by folate deficiency, vitamin B6 or B12 deficiency, renal insufficiency, hypothyroidism, type II diabetes mellitus, pernicious anemia, inflammatory bowel disease, advanced age, climacteric state, carcinoma (particularly involving breast, ovaries, or pancreas), and acute lymphoblastic leukemia, as well as methotrexate, theophylline, and phenytoin therapy.241, 242
The precise mechanisms underlying the thrombogenicity of homocysteine remain unclear. Several diverse mechanisms have been proposed, including endothelial cell desquamation, low-density lipoprotein (LDL) oxidation, promotion of monocyte adhesion to endothelium, and factor V activation and promotion of thrombin generation.241, 242, 243 Homocysteine also enhances platelet aggregation and adhesiveness as well as turnover, presumably as a result of endothelial cell injury.244 One study found that moderate hyperhomocysteinemia does not impair the activation of protein C by thrombin and does not impair the inactivation of factor Va by APC.245
Severe homocysteinemia usually results from homozygous CBS deficiency. The incidence of this disorder is ˜1 in 335,000 live births. Classic symptoms for homozygous patients include premature vascular disease and thrombosis, mental retardation, ectopic lens, and skeletal abnormalities.246 Heterozygous homocysteinemia has been recognized as a disease entity; this disorder may affect 0.3% to 1.0% of the general population.246
Clinical Aspects
Heterozygous carriers of the tlMTHFR mutation have normal plasma homocysteine levels unless folate levels are reduced.247 More important, the majority of case-control studies have not demonstrated an increased VTE risk in homozygous carriers of the tlMTHFR, and the majority of individuals with hyperhomocysteinemia do not have the tlMTHFR polymorphism. Thus, characterization of the tlMTHFR polymorphism is not useful to determine an individual’s VTE risk. VTE risk is most closely related to elevated fasting plasma homocysteine levels, regardless of etiology. Hyperhomocysteinemia (plasma level >18.5 µmol/L) has been associated with a two- to fourfold increased VTE risk.248, 249 It is interesting that, in the Physicians’ Health Study, hyperhomocysteinemia (plasma level above the 95th percentile; 17.2 µmol/L) did increase the risk of idiopathic VTE (relative risk, 3.4) but not the risk of all (transient and idiopathic) VTE.189
The majority of reports linking hyperhomocysteinemia to thrombosis have focused on venous thromboembolic disease. Kottke-Marchant et al. compared 23 patients with documented arterial peripheral thrombosis to age- and sex-matched controls.250 Elevated homocysteine levels (>13 µm/L) conferred an OR of 7.8 for thrombosis. Elevated homocysteine levels were found in 73% of cases versus 28% of controls. Only smoking and homocysteine level were independent risk factors for arterial thrombosis. Currie et al. evaluated homocysteine and cardiovascular risk factors in 66 adult patients with vascular disease. Hyperhomocysteinemia was identified in 29% of patients and was an independent risk factor for the failure of vascular procedures (p = 0.006).251
Laboratory Diagnosis
The initial step in the evaluation of the patient with suspected hyperhomocysteinemia involves measurement of fasting total plasma homocysteine (the sum of nonprotein-bound and proteinbound).241 Many laboratories report homocysteine values in reference to published “normal” ranges such as 5 to 15 µm/L, but, ideally, a local, laboratory-specific normal range should always be established. A normal value in the nonfasting setting does not normally require repeating. Testing 2 to 8 hours after an oral methionine load (100 mg/kg) increases the sensitivity of detecting occult vitamin B6 deficiency and obligate heterozygotes for CBS deficiency,252 but methionine loading is not routinely recommended.253 Vitamin B12 and folate deficiency do not affect post-methionine loading homocysteine values. In patients found to have elevated levels of homocysteine, testing for vitamin B12 deficiency is advocated to avoid missing subclinical deficiency before beginning oral folic acid therapy. Methodologies to measure homocysteine levels have been reviewed.254
Treatment
Folic acid supplementation is the mainstay of effective hyperhomocysteinemia therapy.241 A recent meta-analysis of 1,114 patients enrolled in 12 randomized studies of vitamin supplementation to lower homocysteine levels demonstrated a 25% reduction in homocysteine levels, with similar effects across a dosage range from 0.5 to 5.0 mg daily.255 The usual recommended dose is 0.4 to 1.0 mg daily. Whether patients who are not responsive to one dose benefit from an escalation in dose is unclear. Because patients with subclinical vitamin B12 deficiency may be prone to developing peripheral neuropathy if they receive folic acid supplementation alone, additional treatment with 0.5 mg/day of oral vitamin B12 has been advocated. In the same meta-analysis, an additional 7% reduction of homocysteine levels was noted with vitamin B12 supplementation.255 Vitamin B12 administration results in normalization of homocysteine levels in B12-deficient individuals. In these patients, a monthly intramuscular injection of 200 to 1,000 µg of vitamin B12 is considered adequate replacement. Vitamin B6 supplementation did not appear to have any effect on homocysteine levels. Betaine, a nutritional supplement derived from beets, functions as an alternative methyl donor in the remethylation of homocysteine to methionine. Betaine has been used in individuals with homocystinuria and may facilitate homocysteine reduction in individuals who are not responsive to folate and vitamin B6.256
Thrombotic events in hyperhomocysteinemic patients should be treated as described for other inherited disorders. An additional treatment strategy is to lower plasma homocysteine levels, with the hope of alleviating a risk factor for recurrent thrombosis. However, results of the Heart Outcomes Prevention Evaluation (HOPE)-2 trial and Norwegian Vitamin (NORVIT) trial do not support the utility of homocysteine lowering in patients with arterial disease.257, 258 In patients with a history of coronary artery, cerebrovascular, or peripheral artery disease, or diabetes with at least one risk factor for atherosclerosis, lowering plasma homocysteine levels with folic acid and B vitamins did not reduce risk for the composite endpoint of MI, stroke, or death from cardiovascular causes more than placebo despite a 22% reduction in plasma homocysteine.257 In NORVIT, folic acid and vitamin B12, with or without vitamin B6, did not reduce risk for the composite endpoint of MI, stroke, or death from coronary artery disease in patients with an index MI.258 Other clinical trials have also failed to show a cardiovascular benefit of lowering homocysteine levels.239, 240 Secondary analysis of the HOPE-2 trial regarding effects of vitamin therapy on VTE risk found that there was no significant risk reduction in VTE with vitamin therapy.259 These trials yielding negative results with vitamin therapy raise questions about the utility of testing and treating homocysteinemia.
Increased Factor VIII Activity
It is now appreciated that elevated levels of procoagulant coagulation factors, in addition to deficiencies of natural anticoagulant proteins, are risk factors for VTE. Factor VIII levels >1.5 IU/ml (150%) are associated with a threefold and a sixfold greater relative risk of VTE when compared to levels <1.5 IU/ml (150%) and <1.0 IU/ml (100%), respectively.260 VTE risk is increased 11-fold with levels >200%, but it does not appear to be accentuated by concomitant OCP use.261, 262 Elevated activity levels of factor VIII associated with VTE risk seem to be persistent and not solely attributable to acute-phase response.263 Transiently elevated factor VIII levels associated with acute-phase protein release, though, may in part explain the hypercoagulability associated with inflammatory disorders such as inflammatory bowel disease and cancer. Individuals with plasma factor VIII activity >234% (above the 90th percentile cutoff point for the study population) have a 6.7-fold increased relative risk of recurrent VTE compared to those with activity levels of <120%.42
Because factor VIII is indeed an acute-phase reactant and its levels can be affected by many factors, including blood type and vWF concentration, determination of the true meaning of an elevated factor VIII level in an individual patient with VTE is challenging. In the study by Koster et al., in which blood samples for factor VIII activity were obtained a minimum of 6 months after the VTE, it was impossible to distinguish completely between inherited elevation and postthrombotic, transient elevation of factor VIII.260 Nonetheless, the fact that an increased factor VIII level is prevalent in persons with VTE may, in fact, imply that elevated factor VIII is not only frequent, but also an important risk factor for VTE.214 More recent studies support the concept that elevated factor VIII levels are a significant risk factor in asymptomatic individuals for both arterial and venous thrombosis.264,264a The College of American Pathologists’ consensus conference recommendations are not to routinely measure factor VIII levels in patients with venous or arterial thrombosis.265
Increased Levels of Factors IX, X, XI, and XIII
The Longitudinal Investigation of Thromboembolism Etiology study investigated the possible role of elevated factors IX, X, XI, and XIII in VTE risk266 and found that of these coagulation proteins, only elevated factor XI levels were associated with VTE risk. Elevated factor XI levels have also been linked to stroke267 and coronary disease.268 This effect of elevated factor XI levels may be mediated by increased fibrin formation and decreased fibrinolysis.269
Impaired Endogenous Fibrinolysis
The endogenous fibrinolytic system is comprised of plasminogen, PAs, and antifibrinolytic regulatory proteins. Intravascular plasminogen is converted to the active fibrinolytic enzyme plasmin primarily by t-PA derived from vascular endothelial cells and by urokinase-type PA (u-PA) from leukocytes. The principal physiologic inhibitor of t-PA is PAI-1, whereas plasmin itself is inactivated by circulating and thrombus-bound α2-antiplasmin, and to a lesser extent, α2-macroglobulin. Decreased endogenous fibrinolytic activity as a result of qualitative and quantitative abnormalities of plasminogen, an inadequate release of t-PA in response to vascular injury, and excessive production of PAI-1, such as is found in inflammatory and malignant diseases, can result in impaired endogenous fibrinolysis and accumulation of pathologic thrombus.270
Abnormal fibrinolysis was previously thought to account for a large proportion of patients with inherited thrombosis. However, the discovery of APC-R as a common inherited disorder and a literature review that concluded that a causal relationship between abnormal fibrinolysis and inherited thrombosis had not been clearly demonstrated diminished enthusiasm for routinely evaluating patients with recurrent thrombosis for abnormal fibrinolysis.271 However, newer data from the Leiden Thrombophilia Study indicate that reduced plasma fibrinolytic potential is a risk factor for venous thrombosis,272 and another group identified an elevated level of thrombin-activatable fibrinolysis inhibitor (TAFI) as a risk factor for recurrent venous thrombosis.273 Additionally, elevated levels of α2-antiplasmin are linked to increased risk of MI,274 and elevated levels of PAI-1 are associated with venous thrombosis.275
Plasminogen Deficiency
Quantitative and qualitative abnormalities of plasminogen have been reported in patients with recurrent venous thrombosis.276, 277 Quantitative deficiency is inherited as an autosomal dominant disorder, whereas qualitative plasminogen defects (dysplasminogenemia) are usually inherited as autosomal recessive disorders. Dysplasminogenemia is more common in Japanese subjects, and a single mutation accounts for >90% of cases in this population.278 Two reports have suggested that quantitative plasminogen deficiency279 and dysplasminogenemia280 may not be risk factors for thrombosis, and a recent large series of 50 patients with quantitative plasminogen deficiency found the major clinical manifestation to be ligneous conjunctivitis; venous thrombosis was not observed.281 The College of American Pathologists Consensus Conference on Thrombophilia does not recommend routine assay for plasminogen deficiency in thrombophilia patients.282
Tissue Plasminogen Activator Deficiency
Defective synthesis or release of t-PA from the vessel wall represents a potential mechanism for thrombosis. Plasma t-PA activity is unstable, and for accurate assays, citrated plasma samples must be immediately acidified and red blood cells removed.283, 284 Addition of a platelet activation inhibitor to the blood collection tube may help reduce the release of platelet-derived PAI-1, which can interfere with t-PA activity quantification. A plasminogenchromogenic substrate assay is used to measure t-PA activity; t-PA antigen can be measured by ELISA using citrated plasma. The CAP Consensus Conference on Thrombophilia does not recommend routine assay for t-PA deficiency in thrombophilia patients.282
Increased levels of PAI-1 have been associated with venous or arterial thrombosis. Plasma PAI-1 activity is measured in citrated plasma using a back-titration method with single-chain t-PA.285 Because blood fibrinolytic activity exhibits a diurnal rhythm (decreased fibrinolysis in the morning as a result of peak PAI-1 levels; increased fibrinolysis in the evening as a result of low PAI-1 levels), plasma samples should be obtained at standardized times, such as between 8:00 and 9:00 a.m..286 In addition, because fibrinolytic component levels can be affected by acute-phase changes, patients should not be evaluated for abnormal fibrinolysis until 2 to 3 months after an acute thrombotic event.
In the European Concerted Action on Thrombosis and Disabilities study, an increased incidence of vascular events was associated with higher baseline PAI-1 antigen concentrations (p = .02) and PAI-1 activity (p = .001).53 Elevated levels of PAI-1 may effectively reduce t-PA activity levels, thereby blocking the activation of plasminogen to plasmin and inducing impaired fibrinolysis that may ultimately lead to thrombosis. Theoretically, markedly elevated levels of PAI-1 might also impair attempts at pharmacologic thrombolysis in the setting of acute arterial and venous thrombosis.287 Defects in endogenous fibrinolytic capacity have also been found in young patients with unexplained arterial thromboembolism. Deficient t-PA release was found in 45% of such patients, and elevated levels of PAI-1 were found in 59%.54 Similar defects have been detected in individuals with recurrent venous thrombosis.275 It has been noted that patients who have undergone major surgical procedures might experience a transient “fibrinolytic shutdown” as a result of elevated PAI-1 levels as part of an acute-phase response.288
Avoidance of obesity and associated insulin resistance, correction of hypertriglyceridemia, cessation of smoking, and exercise may improve the innate fibrinolytic status.53, 289, 290 Treatment of hypertension with angiotensin-converting enzyme inhibitors or angiotensin II receptor antagonists has been associated with significantly reduced PAI-1 production.291, 292 The lipid-lowering agent gemfibrozil has also been shown to decrease the PAI-1 synthesis rate.293 Modest alcohol consumption and hormone replacement treatment in postmenopausal women also seem to be beneficial.294, 295
Dysfibrinogenemia
The clinical and biochemical aspects of dysfibrinogens are reviewed in Chapter 53. In general, dysfibrinogens associated with thrombosis have as their molecular defect generation of an abnormal fibrin that is resistant to fibrinolysis. An updated summary of dysfibrinogen molecular abnormalities is presented at www.ISTH.org. Dysfibrinogenemia as a cause of inherited thrombosis is uncommon.
Certain patients with dysfibrinogenemia may have a prolonged PT; more typically, these patients are detected by prolonged thrombin and reptilase times, suggesting a defect in the conversion of fibrinogen to fibrin.296 Confirmation of a dysfibrinogen can be done by simultaneous measurement of functional and immunologic fibrinogen levels. Typically, immunologic levels are found to be higher than levels measured by functional assay. Published data for the healthy population indicate that for fresh plasma samples, the ratio of immunologic to functional fibrinogen ranges from 1.12 to 1.65; ratios >1.65 are suggestive of dysfibrinogenemia.297
Another potentially useful test to evaluate patients for dysfibrinogenemia is the fibrinogen lysis time. This test requires partial purification of the patient’s fibrinogen, followed by clotting and lysis in a standardized assay.298 Dysfibrinogens may also result in a false positive test for fibrin(ogen)-degradation products (FDPs) as assayed by latex agglutination, because residual fibrinogen remains in serum after clotting and is detected by the antibody-coated latex beads. An algorithm for the laboratory diagnosis of dysfibrinogenemia has been reported.299
Thrombomodulin Deficiency
Thrombomodulin complexed with thrombin is required to activate protein C. In theory, thrombomodulin deficiency should result in hypercoagulability. A number of mutations in the thrombomodulin gene have been identified in patients with venous thrombosis and their families.300 Routine laboratory testing for this disorder is not yet available, but an Italian study of thrombosis patients indicated that thrombomodulin deficiency is not a common cause of thrombosis.301 Thrombomodulin gene mutations have been linked to MI.302 In the prospective Atherosclerosis Risk in Communities study, soluble thrombomodulin levels had a graded inverse association with, and were a good predictor of, coronary artery disease.303 The levels correlated in their extreme quartiles with the coagulation activation marker prothrombin fragment 1+2.303
Lipoprotein(a)
Lipoprotein(a) (Lp[a]) is a lipoprotein moiety similar to LDL cholesterol in core lipid composition, and it has apolipoprotein (apo) B-100 as a surface apolipoprotein. In addition, Lp(a) has a unique glycoprotein, apo(a), which is bound to apoB-100. Apo(a) is structurally similar to plasminogen but lacks fibrinolytic activity.304 Lp(a) has both atherogenic and thrombogenic properties. Its thrombogenic effect is derived from stimulation of PAI-1 synthesis, promotion of intercellular adhesion molecule-1 expression by vascular endothelium, inhibition of both plasminogen and t-PA binding to fibrin, inhibition of plasminogen activation by t-PA, and competition with plasminogen for binding sites on endothelial cells and fibrin.305 The cumulative effect is impaired activation of plasminogen to plasmin at the vessel wall, inhibition of fibrinolysis, and increased risk for thrombosis.
The exact mechanisms that control Lp(a) levels are unknown, but genetic factors that regulate hepatic synthesis of apo(a) are likely important. One’s plasma Lp(a) level is stabilized during infancy and maintained throughout life.306 Plasma concentrations >20 mg/dl seem to increase the risk of coronary artery disease, peripheral arterial occlusive disease (PAOD), and stroke.305 Lippi et al. evaluated Lp(a) in 68 patients subjected to vascular and endovascular surgery.307 Significant restenosis or reocclusion occurred in 23 (34%) patients. Lp(a) concentrations were significantly higher in those with restenosis and reocclusion compared with those in the no-restenosis group. Occlusive complications were unlikely to occur in patients with Lp(a) concentrations <5 mg/dl.307 Different isoforms of the apo(a) component of Lp(a) have different atherogenic potential and possibly different thrombogenic potential.
Measurement of Lp(a) should be considered mainly in individuals with atherosclerosis in the absence of classic risk factors, rapidly progressive atherosclerotic lesions despite aggressive risk factor modification, and individuals with acute arterial thrombosis.306, 308 Because it is an acute-phase reactant, an elevated Lp(a) level must be carefully interpreted if drawn shortly after surgery, acute thrombosis, or acute coronary syndromes.309
Patients with elevated Lp(a) benefit most from aggressive LDL cholesterol lowering. Although hepatic 3-methylglutaryl coenzyme A reductase inhibitors (“statins”) may be the ideal agents for LDL lowering and atherosclerotic plaque stabilization, this class of drug does not appreciably lower Lp(a) levels themselves.310 Nicotinic acid (niacin), usually in high doses, is the first-line pharmacologic therapy to lower Lp(a) levels. Reductions up to 38% have been reported.311 Because nicotinic acid therapy may be poorly tolerated, a gradual escalation in dose and pretreatment with aspirin are recommended. In women, estrogen replacement therapy can lower Lp(a) levels by as much as 50%.308
Tissue Factor Pathway Inhibitor
Tissue factor pathway inhibitor (TFPI) is a major anticoagulant, present in plasma, and associated with vascular endothelium. TFPI binds to and neutralizes the TF/factor VIIa complex to inhibit activation of factor X. Low levels of TFPI are a weak risk factor for a first venous thrombosis, as well as recurrent VTE events.312, 313 Recent studies have also linked TFPI deficiency to arterial thrombosis.314
Combined Defects
Compound heterozygosity for two different natural anticoagulant deficiencies is extremely rare, because of the low prevalence of each individual defect. The phenotypic presentation of such compound heterozygotes does not appear to be as severe as it is for the homozygous forms of each deficiency, with the rarely reported patient presenting with VTE of early onset (in the teenage years).98, 214 Because factor V Leiden is the most common inherited thrombophilia, double heterozygosity for factor V Leiden and a natural anticoagulant deficiency has been described in some families.125, 315 In families with both factor V Leiden and protein C deficiency, 73% of compound heterozygotes reported a history of VTE, compared with 31% and 13% of relatives with protein C deficiency only and factor V Leiden only, respectively.125 In families with factor V Leiden and AT deficiency, 92% of double-heterozygous carriers had a history of VTE, as opposed to 57% and 20% of individuals with AT deficiency only and factor V Leiden only, respectively.315
Combined heterozygosity for both the factor V Leiden and prothrombin G20210A mutations, which is estimated to occur in 1 in 1,000 persons, does appear to be associated with an increased relative risk of both first and recurrent VTE.190, 210, 316 In a pooled analysis of eight case-control studies including more than 2,000 patients with VTE, a 2.2% prevalence for the combined mutations was found in patients.190 Double heterozygosity was associated with a 20-fold increased relative risk of VTE.190 The risk of recurrent VTE for heterozygous carriers of both factor V Leiden and PT G20210A also appears to be increased (2.6-fold).210
The prevalence of factor V Leiden carriership in combination with acquired hypercoagulable states has also been studied. Factor V Leiden appears to increase the risk of VTE in patients with antiphospholipid antibodies but is not a prerequisite for the development of VTE in those patients.317 The interaction between factor V Leiden and hyperhomocysteinemia has been a matter of interest since the observation, not corroborated by further studies, that factor V Leiden appeared to be a prerequisite for VTE in individuals from families with classic homocystinuria.318 Nonetheless, two studies on the interaction between mild to moderate hyperhomocysteinemia and factor V Leiden have yielded somewhat different findings. Den Heijer et al. found that the risk of VTE associated with the combination of conditions (2-fold) did not exceed the risk associated with each condition alone (9.5-fold for factor V Leiden, 2.2-fold for hyperhomocysteinemia).248 However, in the large Physicians’ Health Study, the concomitant presence of both conditions did appear to be synergistic.189
Only gold members can continue reading. Log In or Register to continue
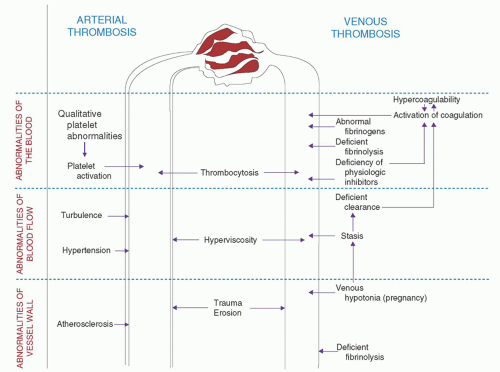

 Clinical Flow Cytometry
Clinical Flow Cytometry



