TABLE 47.1 IMMUNE THROMBOCYTOPENIA | ||||
|---|---|---|---|---|
| ||||
suggesting that the anti-gpIIb/IIIa autoantibody impairs platelet production.25 Megakaryocyte colony formation (colony-forming unit megakaryocyte) is increased in acute ITP.26, 27 In chronic ITP, decreased megakaryocyte colony formation has been reported.28
TABLE 47.2 FEATURES OF ACUTE AND CHRONIC IMMUNE THROMBOCYTOPENIA | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
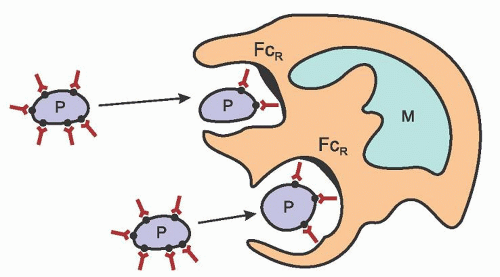 FIGURE 47.1. Antiplatelet antibody-induced destruction of platelets (P) in chronic immune thrombocytopenia. Immunoglobulin binds to platelet-associated antigen, resulting in phagocytosis by macrophages (M). Antiplatelet antibody-coated platelets bind to macrophages through macrophage Fc receptors (FcR). •, platelet membrane antigen; γ, platelet autoantibody. |
TABLE 47.3 CHARACTERISTICS OF PLATELET AUTOANTIBODIES IN PRIMARY IMMUNE THROMBOCYTOPENIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
demonstrate phenotypic and functional abnormalities. Platelet reactive T-cell clones can be identified from the peripheral blood of adults and peripheral blood and spleens of children with chronic ITP, suggesting that autoreactive peripheral T lymphocytes may mediate or participate in disease pathophysiology.53, 78, 79 Adults with ITP often have increased numbers of HLA-DR+ T cells, increased numbers of soluble interleukin-2 receptors, and a cytokine profile suggesting the activation of precursor and mature helper T-cells.79 CD3+ T cells from ITP patients in one DNA microarray study had increased expression of genes involved in cell-mediated cytotoxicity and, in addition, cytotoxic cell-mediated lysis of autologous platelets was shown in active ITP.80
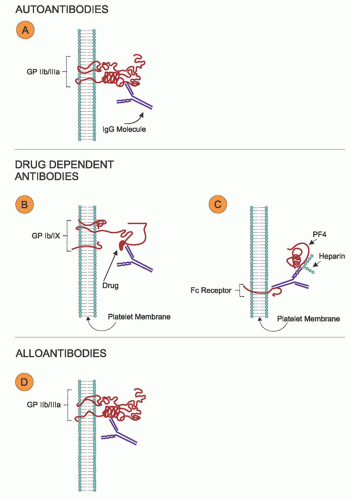 FIGURE 47.2. Types of antibody-mediated platelet destruction. A: Platelet autoantibodies bind to variable external and internal platelet epitopes. B: Quinine/quinidine-dependent antibodies. The antibody target is a complex of drug and glycoprotein (GP) (usually gpIb/V/IX or gpIIb/IIIa). C: Heparin-dependent antibodies. The antigen-antibody complex (target: platelet factor 4 [PF4]/heparin) activates platelets by the binding of immunoglobulin G (IgG) to Fc γRIIA on platelets. D: Platelet alloantibodies bind to platelet tertiary conformational epitopes on the platelet membrane. (Modified from Kelton, reference 40.) |
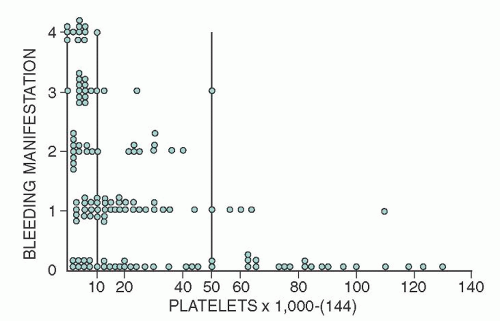 FIGURE 47.3. Bleeding manifestations in relation to platelet count in patients with primary immune thrombocytopenia. Bleeding manifestations (or duration) are graded from 0 to 4, as follows: 0, no bleeding; 1, minimal, resulting from trauma; 2, spontaneous, but self-limited; 3, spontaneous, requiring special attention (e.g., nasal packs); and 4, massive uncontrolled or poorly controlled. (From Lacey and Penner, reference 113.) |
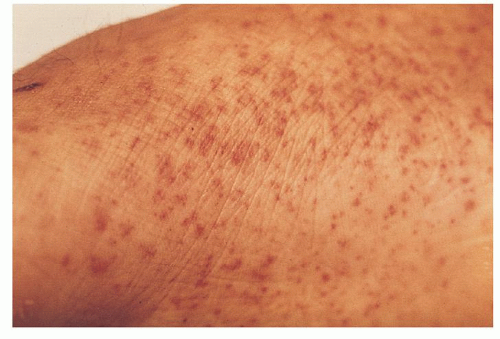 FIGURE 47.4. Petechiae. Pinpoint, nonblanching erythematous capillary bleeding sites are most common in dependent body areas or pressure points. |
accelerated platelet production and the presence of many young forms.137, 138 The changes just summarized are similar to those found in most forms of thrombocytopenia caused by accelerated platelet destruction and are not characteristic or diagnostic of ITP.
fear in the acute form is intracranial hemorrhage, several large studies show that even with low platelet counts (<30,000/µl) life-threatening bleeding and intracranial hemorrhage are rare (<0.5%).166, 167
TABLE 47.4 RECOMMENDATIONS FOR INITIAL TREATMENT OF IMMUNE THROMBOCYTOPENIA (IVIG) PATIENTS WITH PLATELET COUNTS <20,000-30,000/µla | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
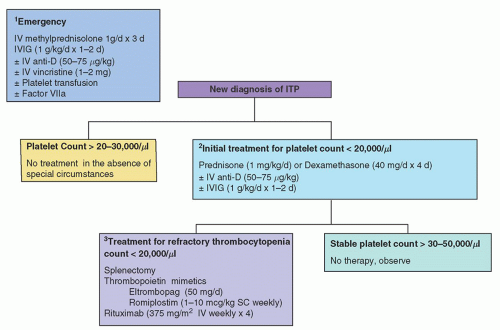 FIGURE 47.5. Therapy of adult immune thrombocytopenia (ITP). (1) Minimal emergency therapy includes intravenous (IV) methylprednisolone and intravenous immunoglobulin (IVIG). Intravenous anti-D and platelet transfusions may be given as needed. All three modalities given prior to transfusions may help preserve platelet longevity in the circulation and repeated or continuous platelet transfusions may be required in urgent situations. (2) Initial treatment of ITP typically consists of steroids (prednisone or dexamethasone) with the goal of attaining a platelet count of >30,000/µl and cessation of bleeding. IVIG or anti-D may be used if steroids are contraindicated or the patient has persistent thrombocytopenia despite steroids. (3) Thrombocytopenia recurs in most adults as corticosteroids are tapered. Treatment options for refractory ITP include splenectomy, thrombopoietin mimetics, and rituximab. (Modified from Cines and Bussel, reference 459.) |
older than age 45 years.115 This risk of fatal bleeding in patients with platelet counts that are chronically <30,000/µl is estimated at 0.4% per year for patients <40 years of age and 13.0% per year for patients >60 years of age.175
TABLE 47.5 THERAPEUTIC AGENTS AND THEIR DOSING SCHEDULES | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


