INTRODUCTION
SUMMARY
Porphyrias are diseases that result from derangements of specific enzymes in the heme biosynthetic pathway that lead to overproduction and accumulation of pathway intermediates and cause neurologic symptoms, photocutaneous symptoms or both. Multiple inherited mutations have been identified in all the porphyrias. However, porphyria cutanea tarda (PCT), which is caused primarily by an acquired deficiency of the fifth enzyme in the heme biosynthetic pathway, specifically in the liver, is usually not associated with a mutation of this enzyme.
Porphyrias can be classified as either hepatic or erythropoietic, depending on the principal site of initial accumulation of excess pathway intermediates. Erythropoietic porphyrias are characterized by childhood onset and a generally stable clinical course. Hepatic porphyrias almost always develop during adult life, and are more variable because of multiple influences of drugs, hormones, and nutritional factors on the heme biosynthetic pathway in the liver.
Porphyrias are also classified as acute or cutaneous. The four acute porphyrias are associated with neurologic manifestations that usually occur as acute attacks. δ-Aminolevulinate dehydratase porphyria (ADP) is an autosomal recessive disorder caused by a deficiency of the second enzyme in the pathway and is the rarest type of porphyria. ADP has been classified as hepatic, but also has erythropoietic features. The three other acute porphyrias, namely acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP), are autosomal dominant hepatic porphyrias, and result from deficiencies of the third, sixth, and seventh enzymes in the pathway, respectively. HCP and VP are also classified as cutaneous, because photocutaneous lesions may develop, especially in VP. AIP is the most common acute porphyria and the second most common porphyria. Disease expression is highly variable in acute porphyrias, and the great majority of individuals who inherit deficiencies of these enzymes remain latent through all or most of their lives. Attacks are produced by factors that increase hepatic heme synthesis, including certain drugs, sex steroid hormones and their metabolites and restriction of dietary calories and carbohydrate. Treatment of acute porphyrias includes glucose loading and hemin infusions, which repress δ-aminolevulinic acid synthase-1, the rate-limiting enzyme of the heme biosynthetic pathway in the liver.
Cutaneous porphyrias are associated with either blistering skin lesions or, in erythropoietic protoporphyria (EPP), with acute nonblistering photosensitivity. Blistering skin manifestations are identical in PCT, HCP, and VP. Similar lesions in congenital erythropoietic porphyria (CEP) are much more severe and often associated with loss of digits and facial mutilation. CEP results from a severe deficiency of the fourth enzyme in the pathway and is inherited in an autosomal recessive fashion. Hemolytic anemia is common, and severe cases may be transfusion dependent, and may even present in utero with fetal hydrops. Hematopoietic stem cell transplantation in early childhood is the most effective treatment.
EPP is the third most common porphyria and the most common in children. It is usually caused by a deficiency of the final enzyme in the pathway. In most families, inheritance of EPP is best described as autosomal recessive, with a severe ferrochelatase mutation inherited from one parent and a low-expression variant allele from the other. X-linked protoporphyria (XLP) has the same phenotype as EPP, but is caused by gain-of-function mutations of δ-aminolevulinic acid synthase-2, which is expressed only in erythroblasts and reticulocytes. Protoporphyrin-containing gallstones may develop in EPP and XLP. An uncommon but potentially life-threatening complication is protoporphyric hepatopathy, which is a result of the cholestatic effects of protoporphyrin, and may require liver transplantation. Sequential marrow transplantation can prevent recurrent hepatopathy in the transplanted liver.
PCT is an iron-related hepatic porphyria that usually begins in middle or late adult life. Activity of hepatic uroporphyrinogen decarboxylase (UROD) is reduced in the presence of iron to approximately 20 percent of normal in PCT by a uroporphomethene inhibitor probably derived from uroporphyrinogen. Multiple susceptibility factors, including use of alcohol, smoking, estrogens, hepatitis C and HIV contribute. HFE (hemochromatosis gene) mutations that cause excess iron absorption are common in PCT. A minority of patients are heterozygous for UROD mutations and are said to have familial PCT. Polyhalogenated aromatic hydrocarbons cause PCT in laboratory animals and occasionally in humans. PCT responds well to treatment by repeated phlebotomy, which reduces hepatic iron, or low-dose hydroxychloroquine or chloroquine, which mobilizes accumulated hepatic porphyrins. Hepatoerythropoietic porphyria is the homozygous form of familial PCT, and is usually a severe disorder that starts in childhood and resembles CEP clinically.
Acronyms and Abbreviations:
ADP, δ-aminolevulinate dehydratase deficiency porphyria; AIP, acute intermittent porphyria; ALA, δ-aminolevulinic acid; ALAD, δ-aminolevulinic acid dehydratase; ALAS, δ-aminolevulinic acid synthase; ALAS1, δ-aminolevulinic acid synthase, housekeeping form; ALAS2, δ-aminolevulinic acid synthase, erythroid-specific form; cDNA, complementary DNA to mRNA template; CEP, congenital erythropoietic porphyria; CPO, coproporphyrinogen oxidase; CPRE, coproporphyrinogen oxidase gene promoter regulatory element; CRIM, cross-reactive immunologic material; CYP, cytochrome P450; EC, enzyme commission; EPP, erythropoietic protoporphyria; FECH, ferrochelatase; HCP, hereditary coproporphyria; HEP, hepatoerythropoietic porphyria; HFE, hemochromatosis gene; HMB, hydroxymethylbilane; IRE, iron-responsive element; IRPs, iron-responsive element binding proteins; NRF-1, nuclear regulatory factor 1; PBG, porphobilinogen; PBGD, porphobilinogen deaminase; PCT, porphyria cutanea tarda; PGC-1α, peroxisomal proliferator-activated cofactor 1α; PPO, protoporphyrinogen oxidase; PXR, pregnane X receptor; SCS-βA, β subunit of ATP-specific succinyl coenzyme A synthetase; UROD, uroporphyrinogen decarboxylase; UROS, uroporphyrinogen synthase; VP, variegate porphyria; XLP, X-linked protoporphyria.
DEFINITION AND HISTORY
The porphyrias are a group of metabolic diseases resulting from derangements, usually of a genetic nature, in the activity of specific enzymes in the heme biosynthetic pathway, leading to overproduction and accumulation of pathway intermediates. Symptoms of these diseases can be neurologic, photocutaneous, or both. The intermediates that accumulate include porphyrins and the porphyrin precursors δ-aminolevulinic acid (ALA) and porphobilinogen (PBG) and their derivatives. Patterns of these substances in plasma, erythrocytes, urine, and feces are characteristic for each porphyria, and are the basis for screening tests and more comprehensive biochemical characterization.
Porphyrias are classified as either erythropoietic or hepatic, depending on the principal site of accumulation of pathway intermediates. The erythropoietic porphyrias are congenital erythropoietic porphyria (CEP), which is very rare, erythropoietic protoporphyria (EPP), the third most common porphyria and the most common in children, and X-linked protoporphyria (XLP), which has the same phenotype as EPP but is less common. Hepatic porphyrias include the acute porphyrias, which cause neurologic symptoms usually in the form of acute attacks, and porphyria cutanea tarda (PCT), which is the most common of the porphyrias, and causes chronic blistering lesions on sun-exposed areas of the skin. The acute porphyrias include ALA dehydratase deficiency porphyria (ADP), acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP). VP, and less commonly HCP, can also cause skin manifestations identical to those in PCT.
A type of porphyria is associated with loss-of-function mutations of seven of the eight enzymes in the heme biosynthetic pathway (Table 58–1 and Fig. 58–1) PCT is primarily caused by an acquired deficiency of the fifth pathway enzyme, with heterozygous mutations of that enzyme contributing in some cases. Gain-of-function mutations of ALAS2, the erythroid form of the first pathway enzyme, cause XLP, whereas loss-of-function mutations of this enzyme cause X-linked sideroblastic anemia (Chap. 59). Table 58–2 summarizes the major clinical and laboratory features of the porphyrias.
Figure 58–1.
Enzymes and intermediates in the heme biosynthetic pathway and the type of porphyria associated with a deficiency of each enzyme (indicated by Ø). Gain-of-function mutation of the erythroid form of ALA synthase is not shown. ADP, ALA dehydratase porphyria; AIP, acute intermittent porphyria; ALA, δ-aminolevulinic acid; ALAD, δ-aminolevulinic acid dehydratase; ALAS, δ-aminolevulinic acid synthase; CEP, congenital erythropoietic porphyria; CoA, coenzyme A; CPO, coproporphyrinogen oxidase; EPP, erythropoietic protoporphyria; FECH, ferrochelatase; HCP, hereditary coproporphyria; PBG, porphobilinogen; PBGD, porphobilinogen deaminase; PCT, porphyria cutanea tarda; PPO, protoporphyrinogen oxidase; SA, sideroblastic anemia; UROD, uroporphyrinogen decarboxylase; UROS, uroporphyrinogen III synthase; VP, variegate porphyria.
| Porphyria* | Affected Enzyme | Known Mutations | Inheritance | Classification | Principal Clinical Features |
|---|---|---|---|---|---|
| X-linked protoporphyria | δ-Aminolevulinic acid (ALA) synthase–erythroid-specific form (ALAS2) | 4 (gain of function) | X-linked recessive | Erythropoietic | Nonblistering photosensitivity |
| δ-Aminolevulinic acid dehydratase porphyria (ADP) | ALA dehydratase (ALAD) | 10 | Autosomal recessive | Hepatic† | Neurovisceral |
| Acute intermittent porphyria (AIP) | PBG deaminase (PBGD) | 273 | Autosomal dominant | Hepatic | Neurovisceral |
| Congenital erythropoietic porphyria (CEP) | Uroporphyrinogen III synthase (UROS) | 36 | Autosomal recessive | Erythropoietic | Neurovisceral |
| Porphyria cutanea tarda (PCT) | Uroporphyrinogen decarboxylase (UROD) | 70 (includes HEP) | Autosomal dominant‡ | Hepatic | Blistering photosensitivity |
| Hepatoerythropoietic porphyria (HEP) | UROD | – | Autosomal recessive | Hepatic† | Blistering photosensitivity |
| Hereditary coproporphyria (HCP) | Coproporphyrinogen oxidase (CPO) | 42 | Autosomal dominant | Hepatic | Neurovisceral; blistering photosensitivity (uncommon) |
| Variegate porphyria (VP) | Protoporphyrinogen oxidase (PPO) | 130 | Autosomal dominant | Hepatic | Neurovisceral; blistering photosensitivity (common) |
| Erythropoietic protoporphyria (EPP) | Ferrochelatase (FECH) | 90 | Autosomal recessive | Erythropoietic | Nonblistering photosensitivity |
| Porphyria | Erythrocytes | Plasma | Urine | Stool |
|---|---|---|---|---|
| XLP | Metal-free and zinc protoporphyrin§ | Protoporphyrin (∼634 nm)‡ | ¶ | Protoporphyrin* |
| ADP | Zinc protoporphyrin | ALA* | ALA, coproporphyrin III | * |
| AIP | Decreased PBGD activity (most cases)* | ALA, PBG* (∼620 nm, some cases)† | ALA, PBG, uroporphyrin | * |
| CEP | Uroporphyrin I; coproporphyrin I | Uroporphyrin I, coproporphyrin I (∼620 nm)† | Uroporphyrin I; coproporphyrin I | Coproporphyrin I |
| PCT and HEP | Zinc protoporphyrin (in HEP) | Uroporphyrin, heptacarboxyl porphyrin (∼620 nm)† | Uroporphyrin, heptacarboxyl porphyrin | Heptacarboxyl porphyrin, isocoproporphyrins |
| HCP | * | ‡(∼620 nm, some cases)† | ALA, PBG, coproporphyrin III | Coproporphyrin III |
| VP | * | Protoporphyrin (∼628 nm)† | ALA, PBG, coproporphyrin III | Coproporphyrin III, protoporphyrin |
| EPP | Metal-free protoporphyrin§ | Protoporphyrin (∼634 nm)† | ¶ | Protoporphyrin* |
A case of CEP reported by Schultz in 1874 was the first description of porphyria in the literature. This case was a 33-year-old man with photosensitivity since age 3 months, anemia, splenomegaly, red-wine-colored urine as a result of a pigment resembling hematoporphyrin, and brown-colored bones at autopsy.1,2 In 1898, T. McCall Anderson described two brothers (ages 23 and 26 years) who most likely had CEP,3 and suffered from hydroa aestivale, with red urine, pruritus and blistering of sun-exposed skin, especially in summer, leading to extensive scarring and mutilation of the ears and nose (see Chap. 58, Fig. 58–5). Using available methods, their urine was also demonstrated to contain a substance related to hematoporphyrin.4 In 1889, Stokvis first described a case of acute porphyria in an elderly woman who developed dark-red urine and later died after taking sulphonal, a drug related to the barbiturates.5
Hans Günther6 published a monograph on porphyrins in 1911 and classified porphyrias into four groups: (1) those that have an acute onset without association with drug ingestion, (2) those that are caused by sulphonal or trional, (3) hematoporphyria congenita, and (4) chronic hematoporphyria. The first two groups correspond to the acute porphyrias, which may present with attacks sometimes related to ingestion of certain drugs, the second group to CEP and hepatoerythropoietic porphyria (HEP), and the fourth to PCT. In 1923, Archibald Garrod proposed the term inborn errors of metabolism for a number of inherited metabolic disorders, including the porphyrias.7
Sachs noted an Ehrlich-positive chromogen that was not urobilinogen in urine of patients with acute porphyria in 1931. In the late1930s, Waldenström noted that excretion of this chromogen was an autosomal dominant trait in AIP families, which he identified as PBG in 1939.8 The classification of porphyrias as erythropoietic and hepatic was proposed in 1954 by Schmid, Schwartz, and Watson.9 An epidemic of hexachlorobenzene-induced PCT in eastern Turkey in 195710,11 provided the foundation for the development of animal models of this disorder using halogenated polyaromatic hydrocarbons.12,13 Strand and coworkers described the enzyme deficiency in AIP for the first time in 1970,14 and Bonkovsky and coworkers first reported treatment of a porphyria patient with hemin in 1971.15 In the past several decades, the enzymes of the heme biosynthetic pathway have been defined in terms of their amino acid composition, genomic and complementary DNA (cDNA) sequences, and crystal structures. Erythroid-specific and housekeeping transcripts have been described for at least four enzymes in the pathway, and progress made in understanding the regulation of heme synthesis in specific tissues, especially the marrow and liver. Multiple mutations have been described in each of the human porphyrias, and some specific treatments introduced.
ETIOLOGY AND PATHOGENESIS
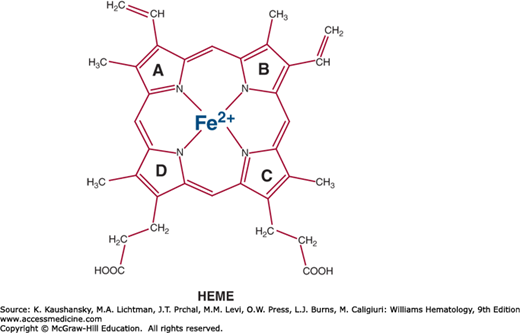
Heme (iron protoporphyrin IX; Fig. 58–2) is essential for all cells and functions as the prosthetic group of numerous hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, cytochromes P450 (CYPs), catalase, peroxidase, tryptophan pyrrolase, and nitric oxide synthase. Approximately 85 percent of heme is synthesized in the marrow to meet the requirement for hemoglobin formation; the remainder is synthesized largely in the liver.16 Most heme synthesized in the liver is required for CYPs, which are located primarily in the endoplasmic reticulum where they turn over rapidly and oxidize a variety of chemicals, including drugs, environmental carcinogens, endogenous steroids, vitamins, fatty acids, and prostaglandins.17
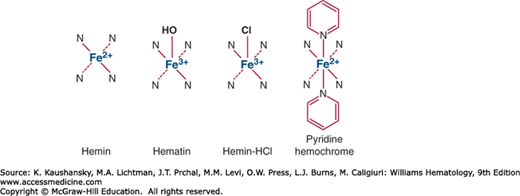
The term heme may refer more specifically to ferrous protoporphyrin IX, and is readily oxidized in vitro to hemin, that is, ferric protoporphyrin IX. Hemin has one residual positive charge and is usually isolated as a halide, most commonly as the chloride. In alkaline solution the halide is replaced by a hydroxyl ion to form hematin (Fig. 58–3). Heme can form further hexacoordinated complexes with nitrogenous bases to form a hemochrome or hemochromogen; for example, pyridine hemochromogen is useful for identification and quantification of heme and hemoproteins. In medicine, hemin is also a generic term for heme preparations used as intravenous therapies for acute porphyrias, such as lyophilized hematin and heme arginate.
The ferrous iron atom (Fe2+) in heme has six electron pairs, of which four are bound to the pyrrolic nitrogens of the porphyrin macrocycle, leaving two unoccupied electron pairs, one above and the other below the plane of the porphyrin ring. In hemoglobin, one of these pairs is coordinated with a histidine residue of the globin chain. The other coordination site in deoxyhemoglobin is protected from oxidation by the nonpolar environment of surrounding amino acid residues, and is available to bind molecular oxygen for transport from the lung to other tissues. To reversibly bind oxygen, the iron in hemoglobin must be in the ferrous state. Methemoglobin (oxidized hemoglobin) that is generated in erythrocytes is continuously reduced to ferrous hemoglobin by the reduced form of nicotinamide adenine dinucleotide–cytochrome b5 reductase–cytochrome b5 system (Chap. 50).
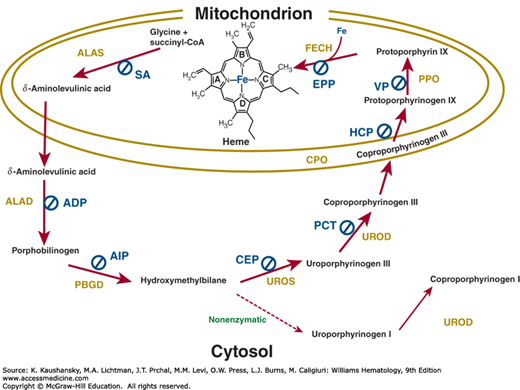
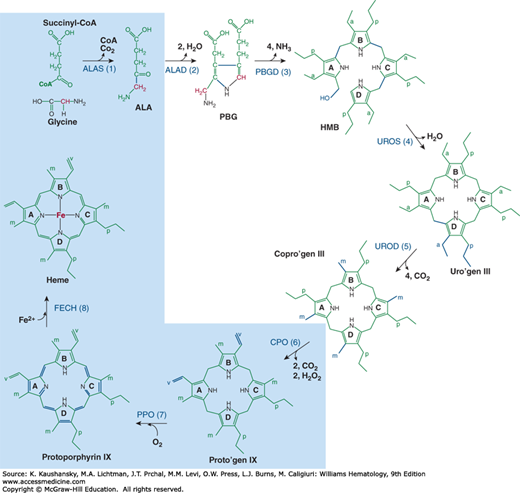
Figure 58–4 shows the enzymatic steps involved in heme biosynthesis in eukaryotic cells. The first and last three enzymes are mitochondrial and the intermediate four are cytosolic. Erythroid heme synthesis occurs in marrow erythroblasts and reticulocytes, which contain mitochondria. Circulating erythrocytes lack mitochondria and no longer synthesize heme. They contain residual cytosolic enzymes of the heme biosynthetic pathway, zinc protoporphyrin and a small amount of metal-free protoporphyrin. These enzyme activities and protoporphyrin decline during the life span of erythrocytes in the circulation.
Figure 58–4.
The heme biosynthetic pathway. The subcellular distribution of the eight enzymes and their substrates and intermediates are shown; enzymes within the light blue shading are located in the mitochondrion, and the others in the cytosol. The substrate positions that are changed are shown in blue, bold lines. ALA, δ-aminolevulinic acid; Copro’gen, coproporphyrinogen; HMB, hydroxymethylbilane; PBG, porphobilinogen; Proto’gen, protoporphyrinogen; Uro’gen, uroporphyrinogen. a, –CH2COOH; p, –CH2–CH2–COOH; m, –CH3; v, –CH=CH2; carbon groups shown in red, carbon atom derived from the α-carbon of glycine; *, location of the α-carbon atom from glycine in the pyrrole ring that undergoes reversion. Step 1, ALA synthase (ALAS); step 2, ALA dehydratase (ALAD); step 3, PBG deaminase (PBGD); step 4, uro’gen III cosynthase (UROS); step 5, uro’gen decarboxylase (UROD); step 6, copro’gen oxidase (CPO); step 7, proto’gen oxidase (PPO); step 8, ferrochelatase (FECH).
δ-Aminolevulinate Synthase (Succinyl Coenzyme A: Glycine C-Succinyl Transferase; Enzyme Commission (EC) 2.3.1.37)
The first enzyme in the heme biosynthetic pathway catalyzes the condensation of glycine and succinyl coenzyme A (CoA) to form ALA (see Fig. 58–4, step 1), and requires pyridoxal 5′-phosphate as a cofactor. δ-Aminolevulinic acid synthase (ALAS) in mammalian cells is localized to the mitochondrial matrix.18 The enzyme is synthesized as a precursor protein in the cytosol and transported into mitochondria. Two separate ALAS genes encode housekeeping (tissue nonspecific) and erythroid-specific forms of the enzyme (ALAS1 and ALAS2, respectively).19 The gene locus for human ALAS1 is at 3p.21, and for ALAS2 it is at Xp11.2.19 The human ALAS2 gene encodes a precursor of 587 amino acids, with an Mr of 64,600 Da. Nucleotide sequences for the ALAS2 and the ALAS1 isoforms are approximately 60 percent similar. No homology is observed between the aminoterminal regions, whereas high homology (approximately 73 percent) is seen after residue 197 of the housekeeping form.21 The two human ALAS genes appear to have evolved by duplication of a common ancestral gene that encoded a primitive catalytic site. Subsequently the DNA sequences were modified to encode gene-specific regulatory regions, functioning mostly at the amino termini.22
The promoter in the human ALAS2 gene contains several putative erythroid-specific cis-acting elements including both a GATA-1 and an NF-E2 binding site.22,23 Both GATA-1 and NF-E2 are erythroid transcription factors that also bind other DNA sites, such as the promoters of the human β-globin, porphobilinogen deaminase (PBGD), and uroporphyrinogen synthase (UROS) genes.24 Thus, expression of ALAS2 is under the regulatory influence of erythroid transcription factors such as GATA-1 and is coordinated with expression of other genes involved in hemoglobin synthesis. Additionally, ALAS2 mRNA contains an iron-responsive element in its 5′-untranslated region,23 similar to mRNAs encoding ferritin and the transferrin receptor (Chap. 42).25 Gel retardation analysis shows that the iron-responsive element in ALAS2 mRNA is functional and suggests that translation of the erythroid-specific mRNA is directly linked to the availability of iron, or heme, in erythroid cells.26
In the liver, synthesis of ALAS1 is induced by a variety of chemicals, including drugs and steroids that increase the demand for hepatic CYPs. Upstream enhancer elements in the ALAS1 gene and certain hepatic CYP genes respond to inducing chemicals and interact with the pregnane X receptor (PXR).27 Hemin represses synthesis of ALAS1 in liver,28 accounting for the beneficial effects of intravenous treatment of the acute porphyrias with hemin. At higher concentrations, heme induces heme oxygenase, resulting in its enhanced catabolism.29 Thus, hepatic heme availability is balanced between the rate of synthesis controlled primarily by ALAS1 and the rate of degradation controlled by heme oxygenase, both of which are regulated by heme at different intracellular concentrations. ALAS1 is also upregulated by the peroxisomal proliferator-activated cofactor 1α (PGC-1α),30 a coactivator of nuclear receptors and transcription factors. Transcriptional regulation of ALAS1 by PGC-1α is mediated by interaction of NRF-1 (nuclear regulatory factor 1) and FOXO-1 (a forkhead family member) with the ALAS1 promoter.31 When glucose levels are low, transcription of PGC-1α is upregulated,32 in turn increasing ALAS1, which might precipitate an attack of acute porphyria in an individual with the appropriate inherited enzyme deficiency. Thus, upregulation of PGC-1α provides an explanation for the induction of acute attacks of porphyria with fasting, as well as the therapeutic value of glucose loading.
Regulation of heme synthesis in erythroid cells is distinct from the liver. ALAS2 expression in erythroid cells is increased during erythroid differentiation when heme synthesis is increased.33,34 Experimentally, ALAS2 is often upregulated by heme, whereas in liver ALAS1 is downregulated by heme. The β subunit of human ATP-specific succinyl CoA synthetase (SCS-βA) associates with human ALAS2 but not with ALAS1, and thereby contributes to heme synthesis in the marrow.35
More than 20 ALAS2 mutations are associated with X-linked sideroblastic anemia (Chap. 59); many are in exon 9, which contains the binding site for pyridoxal 5′-phosphate (K391), and these cases are typically responsive to high doses of pyridoxine. At least one mutant enzyme (D190V) in a patient with pyridoxine-refractory X-linked sideroblastic anemia,36 failed to associate with SCS-βA, whereas other ALAS2 mutants did not have this property. The mature D190V mutant protein, but not its precursor protein, underwent abnormal processing; indicating that appropriate association of SCS-βA and ALAS2 is necessary for functioning of ALAS2 in mitochondria.36 Gain-of-function mutations of ALAS2 have been identified in patients with XLP.37
ALA dehydratase (ALAD) is a cytosolic enzyme that catalyzes the condensation of two molecules of ALA to form the monopyrrole PBG, with removal of two molecules of water (see Fig. 58–4, step 2). The enzyme functions as a homooctamer, and requires intact sulfhydryl groups and zinc for activity. ALAD activity is inhibited by sulfhydryl reagents38 and by lead, which displaces zinc.39 In lead poisoning (Chap. 52), erythrocyte ALAD activity is markedly inhibited, urinary ALA and coproporphyrin excretion increased, erythrocyte zinc protoporphyrin elevated, and neurologic symptoms resemble those seen in acute porphyrias.40 4,6-Dioxoheptanoic acid (succinylacetone) is a substrate analogue and potent inhibitor of ALAD,41,42 and is a byproduct of the enzyme deficiency in hereditary tyrosinemia type I. This substance is found in urine and blood of patients with this disease, who may also have increased ALA and symptoms resembling acute porphyrias.43
Human ALAD mRNA has an open-reading frame of 990 bp, encoding a protein with an Mr of 36,274.44 Sequences known to be essential for enzymatic activity, are those for the active site lysine residues and for the cysteine- and histidine-rich zinc binding sites. The gene for human ALAD is localized to chromosome 9p34.45
Studies using [14C]-ALA have shown that of the two ALA molecules used as substrate, the ALA molecule contributing the propionic acid side is initially bound to the enzyme.38 The tertiary structure of the yeast ALAD has been solved to 2.3-Å resolution, revealing that each subunit adopts a triosephosphate isomerase barrel fold with a 39-residue N-terminal arm. Pairs of monomers then wrap their arms around each other to form compact dimers, and these dimers associate to form a 422 symmetric octamer.46 All eight active sites are on the surface of the octamer and possess two lysine residues (210 and 263). The Lys263 residue forms a Schiff base link to the substrate. The two lysine side chains are close to two zinc binding sites. One binding site is formed by three cysteine residues; the other involves Cys234 and His142.
Although there are no tissue-specific ALAD isozymes, the ALAD mRNA has two splice variants, a housekeeping (1A) and an erythroid-specific (1B) form.45 In both humans and mice, the promoter region upstream of exon 1B contains GATA-1 sites, providing for significant tissue-specific control of these transcripts.47
The human enzyme is polymorphic with two common alleles that occur in three combinations (1–1, 1–2, and 2–2).44 The allele 2 sequence differs from allele 1 only by a G→C transversion of nucleotide 177 in the coding region, resulting in replacement of lysine by asparagine, a more electronegative amino acid.48 ALAD exists primarily as a homooctamer. Mutations associated with ALAD porphyria favor formation of the less active hexamer.49,50
Porphobilinogen Deaminase (Hydroxymethylbilane Synthase; Porphobilinogen Ammonia-Lyase [Polymerizing], EC 4.3.1.8)
The fourth enzyme in the heme biosynthetic pathway catalyzes the deamination and condensation of four molecules of PBG to yield the linear tetrapyrrole hydroxymethylbilane (HMB; see Fig. 58–4, step 3).51 PBG deaminase was previously known as uroporphyrinogen I synthase, and the enzyme activity is commonly measured in the laboratory after converting HMB to uroporphyrin I.
This enzyme has a unique cofactor, which is a dipyrromethane that binds the pyrrole intermediates at the catalytic site until six pyrroles (including the dipyrrole cofactor) are assembled in a linear fashion, after which the tetrapyrrole HMB is released.52 The apo-deaminase generates the dipyrrole cofactor to form the holo-deaminase, and this occurs more readily from HMB than from PBG.53 High concentrations of PBG may inhibit formation of the holo-deaminase.
The gene encoding human PBG deaminase maps to chromosome 11q23→11qter,54 and consists of 15 exons spread over 10 kb of DNA.55 Distinct erythroid-specific and housekeeping isoforms are produced through alternative splicing of two distinct primary mRNA transcripts arising from two promoters.56 The housekeeping promoter is upstream of exon 1 and is active in all tissues, while the erythroid-specific promoter, which is upstream of exon 2, is active only in erythroid cells. The human housekeeping and erythroid-specific enzymes isoforms contain 361 and 344 amino acid residues, respectively.57 Of the additional 17 residues at the N-terminal end of the housekeeping form, 11 are encoded by exon 1, and 6 by a short segment of exon 3 that immediately precedes a methionine codon that initiates translation of the erythroid isoform. Erythroid-specific trans-acting factors, such as GATA-1 and NF-E2, recognize sequences in the erythroid promoter.58 A 1320-bp stretch of perfect identity is present between the erythroid and the nonerythroid PBG deaminase, but with a mismatch in the first exon at their 5′ extremities. An additional inframe AUG codon present 51 bp upstream from the initiating codon of the erythroid cDNA accounts for the additional 17 amino acid residues at the N-terminus of the housekeeping isoform. Accordingly, a splice site mutation at the last position of exon 1, or a base transition in intron 1, in certain patients with AIP results in decreased PBG deaminase expression in nonerythroid tissues including the liver, but not in erythroid cells, because transcription of the gene in erythroid cells starts downstream of the site of the genetic lesion.59
UROS, a cytosolic enzyme, catalyzes the formation of uroporphyrinogen III from HMB. The process involves an intramolecular rearrangement that affects only ring D of the porphyrin macrocycle (see Fig. 58–4, step 4).51 In the absence of this enzyme, HMB spontaneously forms the ring structure uroporphyrinogen I, which, like the III isomer, is a substrate for uroporphyrinogen decarboxylase (UROD). However, because coproporphyrinogen I is not a substrate for coproporphyrinogen oxidase (CPO) the type I porphyrinogen isomers are not further metabolized, and only the type III isomers are precursors of heme.
The UROS cDNA has an open-reading frame of 798 bp, and the predicted protein product consists of 263 amino acid residues, with an Mr of 28,607 Da.20 The amino acid compositions of the hepatic and the purified erythrocyte enzyme are essentially identical, and no tissue-specific isoforms have been described.
The interspecies homology for the UROS proteins is below 10 percent, depending on the number and divergence of the species being compared. However, the crystal structures of uroporphyrinogen III synthase from human and Thermus thermophilus have been solved and are very similar.60,61 The structure supports a mechanism that includes the formation of a spirolactam intermediate by positioning the A and D rings such that the noncatalytic closure, to form uroporphyrinogen I, is not possible.61
UROD is a cytosolic enzyme that catalyzes the sequential removal of the four carboxylic groups of the carboxymethyl side chains in uroporphyrinogen to yield coproporphyrinogen (see Fig. 58–4, step 5). The four successive decarboxylation reactions yield 7-, 6-, 5-, and 4-carboxylated porphyrinogens. Increased amounts of these intermediates can be identified as the corresponding oxidized porphyrins in liver, plasma, urine and stool in human PCT and in laboratory animal models in which hepatic UROD is inhibited. An inhibitor of UROD activity, a partially oxidized substrate molecule,62 is produced in liver of experimental animals in response to halogenated polycyclic aromatic hydrocarbons such as hexachlorobenzene, dioxin, and polychlorinated biphenyls, as well as other compounds able to activate the Ah receptor.63 This porphomethene compound is believed to explain UROD inhibition in human PCT.62 Human UROD is a 42-kDa polypeptide encoded by a single gene containing 10 exons spread over 3 kb and functions as a homodimer.64 The gene has been mapped to chromosome 1p34.65
Although the UROD gene contains two initiation sites, both sites are used with the same frequencies in all tissues, and the gene is transcribed into a single mRNA.66 Recombinant human UROD purified to homogeneity has been crystallized, and its crystal structure was determined at 1.60-Å resolution.67 The purified protein is a dimer with a dissociation constant of 0.1 μM.67 The 40.8-kDa polypeptide forms a single domain with a distorted (β/α)8-barrel fold, and a distinctive deep cleft for the enzyme’s active site is formed by loops at the C-terminal ends of the barrel strands. The protein forms a homodimer with one active-site cleft per monomer located adjacent to its neighbor in the dimer. The structure creates a single extended cleft that is large enough to accommodate two substrate molecules in close proximity. Although both uroporphyrinogen I and III are metabolized by UROD, only the coproporphyrinogen III isomer is further metabolized to heme.68
CPO is located on the outer surface of the inner mitochondrial membrane in mammalian cells.69 The enzyme catalyzes the removal of the carboxyl group and two hydrogens from the propionic groups of pyrrole rings A and B, forming vinyl groups at these positions (see Fig. 58–4, step 6). The enzyme is isomer specific for coproporphyrinogen III, yielding protoporphyrinogen IX (see Fig. 58–4, step 6). The gene for human CPO has been assigned to chromosome 3q12, spans approximately 14 kb, and consists of seven exons and six introns.70 cDNA cloning for this enzyme was first reported in mouse erythroleukemia cells.71 The predicted mouse protein comprises 354 amino acid residues (Mr = 40,647 Da), with a putative leader sequence of 31 amino acid residues. The result is a mature protein consisting of 323 amino acid residues (Mr = 37,225 Da).71 Potential regulatory elements exist in the GC-rich promoter region of the gene, such as six Sp1, four GATA-1, one CACCC site, and the CPO gene promoter regulatory element (CPRE).72 CPRE binds specifically to a CPRE-binding protein, which has a leucine-zipper-like structure and serves as a DNA sequence-specific transcription factor that regulates gene expression.72 Tissue-specific expression of CPO is significant. For example, binding proteins to the Spl-like element, CPRE and GATA-1, cooperatively function in CPO gene expression in erythroid cells. The CPRE-binding protein by itself plays a principal role in basal expression of CPO in nonerythroid cells.73 CPO mRNA increases during erythroid cell differentiation.74
Newly synthesized human CPO contains a 110-amino-acid N-terminal signal peptide,74 which is removed during transport into the intermembrane space of mitochondria, yielding a mature protein of 354 amino acid residues (Mr = 36,842 Da). A five-base insertional mutation in the middle of this presequence has been described in one patient with HCP.75
The penultimate step in heme biosynthesis is the oxidation of protoporphyrinogen IX to protoporphyrin IX, with removal of six hydrogen atoms. This reaction is mediated by the mitochondrial enzyme protoporphyrinogen oxidase (PPO; see Fig. 58–4, step 7). Human PPO cDNA has been cloned.76 The gene is present as a single copy per haploid genome, at chromosome 1q22.77 PPO consists of 477 amino acids with an Mr of 50,800 Da. The deduced protein exhibits a high degree of homology over its entire length to the amino acid sequence of PPO encoded by the HEMY gene of Bacillus subtilis. PPO has been crystallized and the structure shows that the enzyme is a homodimer.78 Sequences required for import into the mitochondria have been identified.79 Expression of PPO is upregulated, approximately fourfold, in the developing erythron from two GATA-1 binding sites located in exon 1.80
The final step of heme biosynthesis is the insertion of iron into protoporphyrin IX. This reaction is catalyzed by the mitochondrial enzyme ferrochelatase (FECH; see Fig. 58–4, step 8). The enzyme uses protoporphyrin IX, rather than its reduced form, as substrate, but requires the reduced ferrous form of iron.81 The gene encoding human FECH has been assigned to chromosome 18q.82 Two FECH mRNA species, approximately 2.5 kb and approximately 1.6 kb in size, are derived from the utilization of two alternative polyadenylation sites in the mRNA. The human FECH gene contains a total of 11 exons and has a minimum size of approximately 45 kb. A major site of transcription initiation is at an adenine, 89 bp upstream from the translation-initiating ATG. The promoter region contains a potential binding site for several transcription factors, Sp1, NF-E2, and GATA-1, but not a typical TATA or CAAT sequence. The transcripts are identical in all tissues examined.
The crystal structure of B. subtilis FECH has been determined at 1.9-Å resolution.83 Subsequently the structure of human FECH was solved and the location of the substrate binding site determined. The enzyme functions as a homodimer and associates with the inside of the inner mitochondrial membrane.83 The mechanism of catalysis has not been identified nor has a function been assigned to the 2Fe-2S cluster that is present in human FECH. Lead inhibits FECH, and a structure of the protein-lead complex has been solved indicating a critical role for the pi helix in catalysis.84 FECH seems to have a structurally conserved core region that is common to the enzyme from bacteria, plants, and mammals.
Tissue-specific aspects of heme synthesis have been studied mostly in erythroid cells and hepatocytes, as the marrow and liver have the greatest requirements for heme. The rate of heme synthesis in the liver is largely regulated by ALAS1 activity. The synthesis of ALAS1, in turn, is under feedback control by heme, which regulates ALAS1 at the levels of transcription, translation, and transfer into mitochondria. Many chemicals, hormones, and drugs increase the synthesis of hepatic CYPs, which increases the demand for heme and lead to induction of ALAS1. In addition, the ALAS1 gene contains upstream enhancer elements that are responsive to inducing chemicals and interact with the PXR. Therefore, ALAS1 and CYPs are subject to direct induction by xenobiotics and certain steroids.27 Chemical exposures that induce hepatic heme oxygenase and accelerate the destruction of hepatic heme, or inhibit heme formation, can also induce hepatic ALAS1.
ALAS2 is not inducible in erythroid cells by drugs that induce ALAS1 in hepatocytes.85 The synthesis of ALAS2 is uninfluenced, or often upregulated, by hemin, at both the transcriptional and the translational levels. Hemin treatment of marrow cultures increases erythroid colony-forming units,86 whereas hemin treatment of hepatocytes inhibits synthesis of ALAS1 and CYPs. An additional distinct difference in these ALAS isoforms is that SCS-βA associates with ALAS2 but not with ALAS1, suggesting a tissue-specific difference in mitochondrial transport of these isoforms.
ERYTHROPOIETIC PORPHYRIAS
There are two major erythropoietic porphyrias in humans. CEP is caused by mutations of the UROS gene. It is one of the least-common porphyrias, but is well known as a result of its long history and the severe photomutilation of exposed areas such as the face and fingers that is a dramatic feature in many cases (Fig. 58–5). EPP, which is caused by FECH mutations, is the third most common porphyria, and the most common in children, but was not well described until 1965. XLP is much less common, has the same phenotype as EPP, but normal FECH activity. In 2008, the discovery of gain-of-function mutations in the last exon of ALAS2 provided an explanation for the increased level of erythrocyte protoporphyrin seen in this type of protoporphyria.37 Characteristics in most patients with erythropoietic porphyrias that are distinct from the hepatic porphyrias include childhood onset, stable symptoms and levels of porphyrins over time, and severity largely determined by genotype rather than factors that affect the heme pathway, primarily in the liver. Substantial increases in erythrocyte zinc protoporphyrin in ADP and homozygous forms of other hepatic porphyrias, such as HEP (the homozygous form of familial PCT), AIP, HCP, and VP, suggest that an erythropoietic component may be important in these conditions.87
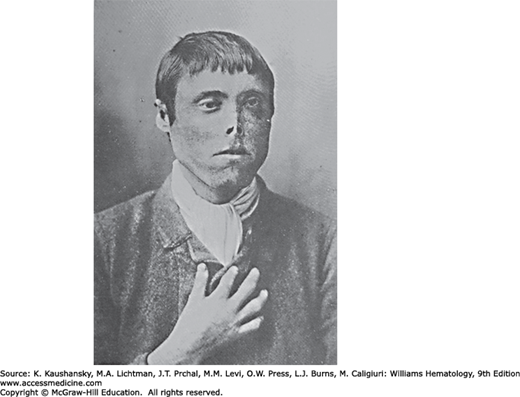
Figure 58–5.
A 23-year-old Scottish fisherman with congenital erythropoietic porphyria and scarring and mutilation of the face, ears, and digits as a consequence of repeated sun exposure. He was described in 1898 as having red urine containing excess porphyrins and “hydroa aestivale,” because the symptoms, which began at age 3 years, worsened in early summer. A 26-year-old brother was similarly affected. (Reproduced from Anderson TM.3)
CEP is caused by a deficiency of UROS (see Fig. 58–4, step 4), is an autosomal recessive condition, and is also known as Günther disease. It results in accumulation and excretion of isomer I porphyrins, especially uroporphyrin I and coproporphyrin I (see Table 58–1 and Fig. 58–1). Characteristic manifestations of CEP include chronic, severe photosensitivity and hemolytic anemia evident in early childhood. Atypical presentations include milder disease that resembles PCT, and onset during adult life often in association with a myeloproliferative disorder.88 Early case descriptions of CEP appeared in 1874 and 1898,3 and approximately 130 cases were reported up to 1997.89 However, some of these patients may have had HEP, which has very similar clinical features. Perhaps the most well-known patient was Mathias Petry, who survived until age 34, and beginning in 1915, worked with the porphyrin chemist Hans Fisher, providing samples for early studies of porphyrin chemistry.90
The uroporphyrinogen III synthase defects in CEP are remarkably heterogeneous at the molecular level, with at least 46 different mutations of the UROS gene, and one GATA-1 mutation reported as of this writing.87 The UROS mutations include deletions, insertions, rearrangements, splicing abnormalities, and both missense and nonsense mutations.
The missense mutations are well distributed throughout the gene. Of the 12 single-base substitutions, four (T228M, G225S, A66V, A104V) were hotspot mutations, occurring at CpG dinucleotides.91 The identification of a mutation that altered the penultimate nucleotide in exon 4, resulting in an E81D mutation, also produced exon skipping on approximately 85 percent of the transcripts from that allele. With the exception of V82F, all CEP missense mutations occurred in amino acid residues that are conserved in both the mouse and the human enzyme.
Genotype–phenotype correlation in CEP was studied by prokaryotic expression of mutant UROS cDNAs. Mean activities of the mutant enzymes expressed in Escherichia coli ranged from 0 to 36 percent of the activity expressed by the normal cDNA. The majority of the mutant cDNAs expressed polypeptides with no enzymatic activity. However, V82F, E81D, A66V, A104V, and V99A showed 36, 30, 15, 8, and 6 percent enzyme activity, respectively, compared with the normal control. A66V and V82F were thermodynamically unstable mutants.91 Homoallelism for C73R, the most common mutation, found in five patients, was associated clinically with the most severe phenotypes, such as hydrops fetalis and transfusion dependency from birth.
Porphyrins in their oxidized state are reddish, fluorescent, and photosensitizing, whereas porphyrin precursors and the reduced porphyrinogens are colorless and nonfluorescent. Most marrow normoblasts in CEP display marked fluorescence as a result of porphyrin accumulation (located principally in the nuclei, probably because of fixation artifact).92 Anemia and the excess production and excretion of porphyrins is largely accounted for by ineffective erythropoiesis in the marrow. Porphyrin concentrations are also increased in circulating erythrocytes, and intravascular hemolysis may result from exposure to light in the dermal capillaries, causing erythrocyte damage and lysis or uptake by the spleen. Splenomegaly is very common in CEP and is presumed to be secondary to the hemolytic process. The excess porphyrins that are produced by the marrow or released by hemolysis are transported in plasma to the skin, leading to photosensitivity.
Severe cutaneous photosensitivity is noted soon after birth in most cases. The disease may be recognized even earlier as a cause of hydrops fetalis. Phototherapy for hyperbilirubinemia may cause severe cutaneous burns and scarring in newborns with unrecognized CEP. Brown staining of the teeth by porphyrins (erythrodontia) is evident when the teeth erupt. Blistering and scarring resemble those found in PCT, but are usually much more severe, reflecting the much higher plasma porphyrin levels observed in CEP. Some cases are relatively mild, and can closely mimic PCT. Late-onset cases are often associated with myeloproliferative disorders, with expansion of a clone of erythroid cells bearing a somatic mutation and displaying UROS deficiency.88
Subepidermal bullous lesions are characteristic, and progress to crusted erosions that heal with scarring and areas of hyper- and hypopigmentation. Also common is hypertrichosis, which is sometimes severe, and alopecia. Loss of facial features and digits are common and result from recurrent blisters, infection, and scarring. Fingers may be shortened and tapered as a consequence of scarring and contraction of the skin during childhood growth. Erythrodontia, with brown staining and red fluorescence of the teeth under long-wave ultraviolet light is characteristic, and results from deposition of porphyrins in the developing deciduous and permanent teeth in utero. Porphyrins are also deposited in bone. The skeleton is also affected by expansion of the marrow, leading to pathologic fractures, vertebral compression, short stature, and osteolytic and sclerotic lesions. Vitamin D deficiency resulting from avoiding sunlight might also contribute.
Anemia may be severe and lead to transfusion dependence in the more severe cases. Uncorrected anemia can increase erythropoiesis, which, in turn, is a stimulus to porphyrin production by the abnormal erythropoietic cells in the marrow. Erythrocytes exhibit polychromasia, poikilocytosis, anisocytosis, and basophilic stippling, and reticulocytes and nucleated red blood cells are increased.93
CEP may be suspected even before birth if a sibling is known to have CEP. However, the family history is often negative. CEP should be suspected as a cause of hydrops fetalis, as the disease can be diagnosed and treated in utero. Aspirated amniotic fluid is dark brown in color and contains large amounts of porphyrins. The diagnosis of CEP is often made after birth when pink to dark-brown staining of the diapers, with red fluorescence under long-wave ultraviolet light, is noted. Cutaneous vesicles and bullae on sun-exposed areas may be severe, with scarring.
Urinary porphyrin excretion is markedly increased, and often in the range of 50 to 100 mg/day (normal: up to ~0.3 mg/day). Uroporphyrin I and coproporphyrin I account for most of the increase, although the III isomers and hepta-, hexa-, pentacarboxylate porphyrins are also increased. Fecal porphyrins are increased, and are predominantly coproporphyrin I. Plasma total porphyrins are markedly increased as well, with a pattern of individual porphyrins similar to that in urine. Markedly increased erythrocyte porphyrins are predominantly uroporphyrin I and coproporphyrin I, although protoporphyrin IX may predominate especially in milder cases.
CEP must be distinguished by biochemical testing from other causes of blistering skin lesions. HEP can present with photosensitivity in early childhood. Mild cases of CEP may be misdiagnosed as PCT.
The diagnosis should be confirmed in all cases by DNA studies, which can identify causative mutations in almost all cases. This is especially important for genetic counseling and for prenatal diagnosis in subsequent pregnancies. Demonstration of a GATA-1 mutation in one case, illustrates that on occasion a genetic defect outside the heme biosynthetic pathway can cause CEP.93
Patients should be advised that to avoid severe scarring and loss of facial features and digits it is essential to avoid sunlight, trauma to the skin, and infections. Topical sunscreens that block long-wave ultraviolet light (ultraviolet A light) and oral treatment with β-carotene are somewhat helpful,94 but are marginally beneficial in most cases. Erythrocyte transfusions are essential in patients with severe anemia.95 Transfusions to maintain the hematocrit above 35 percent, with an iron chelator to avoid iron overload, has been beneficial in some cases.96 Hydroxyurea to reduce erythropoiesis and porphyrin production may also be considered.97 Splenectomy has provided short-term benefit. Oral charcoal reportedly was quite effective in one patient,98 and ascorbic acid and α-tocopherol improved anemia in another.89 Unaffected infants born to mothers with CEP may have erythrodontia as a result of exposure to maternal porphyrins before birth.99
Hematopoietic stem cell transplantation is the treatment of choice when a suitable donor is available, especially for young patients.100 When transplantation is successful, there is marked clinical improvement and reduction in porphyrin levels, even if these are not completely normalized. Gene therapy is being explored using retroviral and lentiviral vectors and hematopoietic stem cells from patients with CEP.101,102
EPP is caused by a partial deficiency of FECH (see Fig. 58–4, step 8) activity, which results in the accumulation of the substrate protoporphyrin in the marrow. XLP has the same phenotype but is much less common, and is a result of ALAS2 gain-of-function mutations (ALAS; see Fig. 58–1).37 EPP and XLP are characterized by onset of nonblistering cutaneous photosensitivity in early childhood. EPP is the most common porphyria in children and the third most common in adults. Reported prevalence varies between 5 and 15 cases per 1 million individuals.103,104,105 Protoporphyric hepatopathy is a potentially fatal complication estimated to occur in less than 5 percent of patients.
In most families, EPP is best described as an autosomal recessive disease, in which a severe FECH mutation is inherited from one parent and a low-expression (hypomorphic) FECH allele from the other. More than 75 different severe mutations, including nonsense, missense, and splice-site mutations, and deletions, insertions, and rearrangements have been described. Splicing mutations are most common. Recombinant human FECH, when engineered to have individual exon skipping for exons 3 through 11, lacks significant enzyme activity when expressed in E. coli and almost all such variants lacked the [2Fe-2S] cluster.106
EPP was usually described in the past as an autosomal dominant disorder, with variable penetrance. However, it was noted that EPP patients have only 30 percent or less of normal FECH activity, rather than 50 percent, which would be expected in an autosomal dominant condition. It was then shown that, in addition to a severe FECH mutation, a low-expression (hypomorphic) intronic polymorphism (a –23C→T transition) is found in the other FECH allele of patients with EPP, which is inherited from the other parent.107,108,109 This transition favors the use of a cryptic acceptor splice site 63 bases upstream of the normal splice site. The aberrantly spliced mRNA contains a premature stop codon and is degraded by a nonsense-mediated decay mechanism.109 The result is a lower steady-state level of wild-type FECH mRNA. Coinheritance of the hypomorphic allele in trans to a loss-of-function mutant allele was found in 98 percent of French cases with EPP,110 and with a similar frequency in South African patients.105 The frequency of the IVS3–48C hypomorphic allele is common in the white population, and by itself has no phenotype. Its frequency varies widely in different populations and relates to the observed differences in the prevalence of EPP.103,104,105
Other underlying genetic mechanisms must be considered in newly identified EPP families. In a few families, a severe FECH mutation, at least one of which must produce some FECH enzyme, is inherited from each parent and the hypomorphic allele is not present. Interestingly, EPP in such families is sometimes associated with seasonal palmar keratoderma, unusual neurologic symptoms, less-than-expected increases in erythrocyte protoporphyrin and absence of liver dysfunction.111
XLP was first perceived as a variant form of EPP in which FECH mutations were absent. After family studies suggested sex-linked inheritance, gain-of-function mutations of ALAS2 (the only heme pathway enzyme found on the X chromosome) were discovered.37 This is the only porphyria caused by mutations of ALAS, the first enzyme in the pathway.
EPP can develop late in life in patients with clonal hematologic disorders and expansion of a clone of hematopoietic cells with mutations of a FECH allele.112,113 For example, a patient with a myeloproliferative disorder later developed severe EPP because of clonal expansion of a cell of erythropoietic lineage with a FECH deletion and the IVS3–48C/T polymorphism, and died of EPP-induced liver disease.114
Marrow reticulocytes are thought to be the primary source of the excess protoporphyrin in EPP.115,116 Most of the excess erythrocyte protoporphyrin in circulating erythrocytes is found in younger cells as metal-free protoporphyrin (i.e., not complexed with zinc), in contrast to other conditions associated with increased erythrocyte protoporphyrin content. Metal-free protoporphyrin declines much more rapidly with red cell age than it does zinc protoporphyrin.115,116 Metal-free protoporphyrin, but not zinc protoporphyrin, is released from erythrocytes following solar irradiation, which may explain why lead intoxication and iron deficiency, which are associated with elevated erythrocyte zinc protoporphyrin levels, are not associated with photosensitivity.117 Excess metal-free protoporphyrin enters plasma from reticulocytes, as well as from circulating erythrocytes, and is taken up by hepatocytes, excreted in bile and feces, and may undergo enterohepatic recirculation. Hepatocytes may also provide a limited additional source of excess protoporphyrin in this disease.
Light-excited protoporphyrin in EPP generates free radicals and singlet oxygen,118 which in EPP can lead to peroxidation of lipids119 and crosslinking of membrane proteins. Skin irradiation in EPP patients leads to complement activation and polymorphonuclear chemotaxis, which contributes to the development of skin pathology.120 Skin histopathology is not specific but may include thickened capillary walls in the papillary dermis surrounded by amorphous hyaline-like deposits, immunoglobulin, complement, and periodic acid-Schiff–positive mucopolysaccharides.121 Basement membrane abnormalities are less marked than in other forms of porphyria.122
Protoporphyric hepatopathy is a feared complication that develops in less than 5 percent of patients, and is attributed to the cholestatic effects of excess protoporphyrin presented to the liver. This complication may begin with chronic abnormalities in liver function tests and then progress rapidly as a vicious cycle of increasing protoporphyrin levels in plasma and erythrocytes and worsening liver function and photosensitivity. Hepatopathy is sometimes precipitated by another cause of liver dysfunction such as viral or alcoholic hepatitis. Protoporphyrin is cholestatic, and can form crystalline structures in hepatocytes and impair mitochondrial function, leading to decreased hepatic bile formation and flow.123,124 Accumulated protoporphyrin may appear as brown pigment in hepatocytes, Kupffer cells, and biliary canaliculi, and these deposits are doubly refractive with a Maltese cross appearance under polarizing microscopy.125 DNA microarray studies in explanted livers of patients with hepatopathy revealed significant changes in expression of several genes involved in wound-healing, organic anion transport, and oxidative stress.126
Photosensitivity is present from early childhood in almost all cases. Parents may observe that an affected infant cries and develops skin swelling and erythema when exposed to sunlight. Although EPP is the most common porphyria in children, there is often considerable delay in diagnosis.
Cutaneous photosensitivity in EPP is acute and nonblistering, which is distinctly different from the more chronic, blistering skin manifestations of the other cutaneous porphyrias. Table 58–3 tabulates symptoms in a series of 32 patients with EPP. Skin symptoms are usually worse during spring and summer and affect light-exposed areas, especially of the face and hands. Characteristically, stinging or burning pain develops within 1 hour of sunlight exposure, and if exposure continues is followed by erythema and edema—described as solar urticaria, sometimes with petechiae, and less commonly purpura. Blistering and crusted lesions are uncommon. Artificial lights may contribute to photosensitivity.127 Patients typically avoid sunlight and may display no objective cutaneous signs. Repeated light exposure can lead to chronic changes including leathery hyperkeratotic skin especially on the dorsa of the hands and finger joints, mild scarring, and separation of the nail plate (onycholysis).
Mild anemia with microcytosis, hypochromia, reduced iron stores, but usually normal serum iron, and serum transferrin receptor-1, is a common feature of EPP,115,128,129,130,131 but there is little evidence for impaired erythropoiesis or abnormal iron metabolism,129,130 and hemolysis is absent or very mild.131,153 Iron accumulation in erythroblasts and ring sideroblasts have been noted in marrow in some patients.132 Findings in XLP are similar. Also, iron is proposed to have a role in splicing of FECH mRNA, where decreased iron leads to an increase in incorrect splicing of the mRNA.133 Binding of iron-responsive elements binding proteins (IRPs) to the 5′-iron-responsive element (IRE) in ALAS2 mRNA, in low iron conditions, prevents translation of ALAS2 mRNA. When iron is supplemented, the IRPs no longer have high affinity for the ALAS2 5′-IRE, leading to increased translation, import into the mitochondria, and enhanced production of ALA.134
Precipitating factors that are important in the hepatic porphyrias do not appear to play an important role in EPP. Although more long-term followup studies are needed, porphyrin levels and symptoms typically do not change over time, unless liver dysfunction develops. Concurrent iron deficiency or other marrow problems might also lead to further increases in porphyrin levels and photosensitivity. Pregnancy is reported to lower erythrocyte protoporphyrin levels somewhat and increase tolerance to sunlight.135
Neurovisceral manifestations are absent in uncomplicated EPP. Patients with severe protoporphyric hepatopathy may develop a severe motor neuropathy similar to that seen in the acute porphyrias.136 Autosomal recessive EPP associated with palmar keratoderma has also been associated with unexplained neurologic symptoms.111
Gallstones containing large amounts of protoporphyrin are common, and may require cholecystectomy at an unusually early age.137 Liver function and liver protoporphyrin content are usually normal in EPP. Protoporphyric hepatopathy, which is the most life-threatening complication of EPP, results from the cholestatic effects of protoporphyrin presented in excess amounts to the liver. It can be the major presenting feature of EPP,138 and may be chronic or progress rapidly to death from liver failure. Unnecessary surgery for suspected biliary obstruction can be detrimental and should be avoided.124 Operating room lights during liver transplantation or other surgery, especially in patients with hepatopathy, can cause marked photosensitivity with extensive burns of the skin and peritoneum and photodamage of circulating erythrocytes.139
Painful, nonblistering photosensitivity suggests the diagnosis. A substantial elevation of erythrocyte protoporphyrin is expected, but is not specific, as erythrocyte zinc protoporphyrin is predominantly increased in conditions such as homozygous porphyrias (other than most cases of CEP), iron deficiency, lead poisoning, anemia of chronic disease,140 hemolytic conditions,141 and many other erythrocyte disorders. A unique finding in EPP is increased erythrocyte protoporphyrin with a predominance of metal-free rather than zinc protoporphyrin. This occurs because FECH, which can utilize metals in addition to iron, catalyzes the formation of zinc protoporphyrin, and this activity is deficient in EPP. Because FECH is not deficient in XLP, erythrocytes contain increased amounts of both zinc and metal-free protoporphyrin, although the latter still predominates in most cases.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


