INTRODUCTION
SUMMARY
Gaucher disease and Niemann-Pick disease are the two lipid storage disorders that are most likely to be encountered by the hematologist because both may cause hepatosplenomegaly and cytopenias.
Gaucher disease is a common autosomal recessive lipid storage disorder, with an increased prevalence among Ashkenazi Jews, in whom the estimated birth occurrence is 1 in 850. Deficiency of the enzyme β-glucocerebrosidase results in accumulation of the sphingolipid glucocerebroside in the cells of the macrophage-monocyte system. Patients with the most prevalent form, type 1, have no primary neuronopathic symptoms, whereas there is involvement of the central nervous system in type 2 and type 3. Diagnosis of Gaucher disease depends on demonstration of decreased enzymatic activity of β-glucocerebrosidase combined with identification of mutations in the β-glucocerebrosidase gene at the DNA level, usually with elevation of biomarkers, such as chitotriosidase as ancillary confirmation and means of followup. Disease manifestations include hepatosplenomegaly, thrombocytopenia, anemia, osteopenia/osteoporosis with pathologic fractures and osteonecrosis, and, less commonly, pulmonary infiltration. Many patients, especially those homozygous for the common N370S mutation, are putatively protected against neurologic involvement, albeit there is evidence of a genetic risk factor for Parkinson disease. Generally, many patients with type 1 may be asymptomatic or so mildly affected that they may not present until their fifth or sixth decade and do not require disease-specific therapy, whereas for those with more severe signs and symptoms, enzyme replacement therapy (currently three infusible enzymes) is available. Substrate reduction therapy is an oral modality but is associated with a more problematic safety profile. Pharmacological chaperones and oral enzyme are being tested.
Niemann-Pick disease is a heterogeneous group of autosomal recessive disorders. Type A and type B result from deficiency of the enzyme sphingomyelinase, whereas type C results from mutations in the NPC1 or NPC2 gene, which appears to be involved in cholesterol trafficking and resulting in accumulation of cholesterol as well as sphingomyelin. Type A is a lethal infantile form with marked progressive neurologic involvement. Type B is a later-onset form with no neurologic involvement but hepatosplenomegaly in many patients. Patients with type C disease manifest progressive neurologic involvement and hepatosplenomegaly, but may survive into adulthood. The marrow of these patients contains typical foam cells with small droplets in the cytoplasm and sea-blue histiocytes. Substrate reduction therapy was approved for patients with type C disease in 2008 in Europe; pharmacologic chaperone therapy is being attempted.
Fabry disease, Wolman/cholesteryl ester storage disease (CESD), and GM1-gangliosidoses are other lipid storage diseases characterized by hepatosplenomegaly; GM2-gangliosidosis by hepatomegaly only. CESD patients may result in anemia and have sea-blue histiocytes. They are usually not cared for by hematologists and will not be discussed in this chapter.
Acronyms and Abbreviations
cDNA, complementary DNA; ERT, enzyme replacement therapy; MRI, magnetic resonance imaging; PC, pharmacologic chaperone; SRT, substrate reduction therapy.
DEFINITION OF GLYCOLIPID STORAGE DISEASES
The glycolipid storage diseases are hereditary disorders in which one or more tissues become engorged with specific lipids, because of deficiencies of the lysosomal enzymes required for hydrolysis of one of the glycosidic bonds. Figure 72–1 shows the catabolic pathway of glycosphingolipids and lists the diseases that are involved in impaired degradation because of specific enzyme deficiencies. The type of lipid and its tissue distribution have a characteristic pattern in each disorder. This chapter deals mainly with Gaucher disease, in which glucocerebroside is stored. It is a common lysosomal storage disorder and also the one with the most hematologic features. The second storage disorder with some hematologic features is Niemann-Pick disease, in which the accumulated material is sphingomyelin and/or cholesterol. The remaining lysosomal diseases (Fabry disease, Wolman/cholesteryl ester storage disease, and GM1– and GM2-gangliosidoses), in which there is hepatosplenomegaly but few hematologic abnormalities, are not reviewed in this chapter.
Figure 72–1.
The catabolic pathways of selected glycosphingolipids involved in some of the glycolipid storage diseases. Solid squares depict the blocked pathways caused by specific inherited deficiencies of enzymes, which give rise to the accumulation of the respective substrates. The names of the various diseases are shown above the names of the deficient enzymes. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
GAUCHER DISEASE
Gaucher disease was first described by P.C.E. Gaucher in 1882, who thought that the large splenic cells of a young woman seen postmortem were evidence of a primary neoplasm.1 The term Gaucher disease appeared first in 1905, when the autosomal recessive genetic nature of the disorder was elucidated.2 In 1934, it was shown that glucocerebroside is the storage material in Gaucher disease,3 and in 1965, the primary defect was recognized as a deficiency of glucocerebrosidase resulting in an impairment of degradation of glucocerebroside.4,5 Enzymatic purification ultimately led to the cloning of the gene in 1985,6,7 unraveling of its structure, and identification of many glucocerebrosidase mutations.8 Disease-specific enzyme replacement therapy (ERT) was first introduced in 1991.9
Gaucher disease is inherited as an autosomal recessive disorder. Although panethnic, type 1 is most common among the Ashkenazi Jews, with a carriership prevalence of 1 in 17 and an expected frequency of the disease in 1 in 850 livebirths.10 Two distinct forms of Gaucher disease, type 3b and type 3c, are also relatively common in Norrbottnia in northern Sweden,11 and near the Palestinian town of Jenin, respectively.12 In the general population, the estimated frequency (based on large-scale neonatal screening projects in three countries is in the range of 1 in 50,000 to 1 in 100,000 persons.13
The high prevalence of at least two Gaucher mutation, N370S and 84GG (and possibly R496H14 and others), among Ashkenazi Jews, and the existence of other lysosomal diseases within this ethnic group, may reflect, in addition to a founder effect, a selective advantage. However, a selective advantage because of greater resistance to tuberculosis15 or superior intelligence16 has not been proven. Animal studies suggest that the selective advantage may be the higher circulating serum levels of glucocerebroside that have antiinflammatory and beneficial immunomodulary effects.17
During normal growth, development, and senescence, parts of or whole cells are continually replaced. Breakdown of complex constituents of cells requires sequential enzymatic degradation. Such degradation occurs largely in secondary lysosomes, organelles formed by the fusion of primary lysosomes with phagocytic vacuoles containing ingested material.
Gaucher disease is the result of a hereditary deficiency in the activity of a lysosomal enzyme, glucocerebrosidase, required for glycolipid degradation. The reduced activity of glucocerebrosidase results in accumulation of glucocerebroside in macrophages engorgement with glucocerebroside induces increased cell size and cytoplasmic striations, leading to the formation of “Gaucher cells” (see Fig. 72–1). Inherent in subsequent lysosomal dysfunction is a dysregulation of metabolites and the consequent lack of coordination of cellular metabolism. These changes may explain the elaboration of various cytokines and other biomarkers because of engorgement of macrophages.
Accumulating evidence indicates that in addition to the glucocerebrosidase enzyme (GBA1), a second, nonlysosomal glucocerebrosidase, GBA2, a cytosolic protein that tightly associates with cellular membranes, may be integral to the pathogenesis of Gaucher disease, affecting its phenotype by potentially interacting with GBA1.18,19,20
In rare instances, severe forms of Gaucher disease are associated with deficiency of saposin C, a heat-stable glucocerebrosidase co-factor.21,22
The glucocerebrosidase gene is located on chromosome 1q21. A pseudogene, with 96 percent sequence homology, has been identified approximately 16 kb downstream from the functional gene. Nearly 300 point mutations causing Gaucher disease have been described8,23; most are point mutations, missense, nonsense, frameshift, and splice-site mutations, but there are also insertions, deletions, and recombinant alleles. Some mutations result from recombinant events between the functional gene and its pseudogene.8 Since 2000, approximately 20 of these mutated enzymes have undergone crystallography showing the divergence of ligands in the active site and with various degrees of glycosylation.23
Among Ashkenazi Jews, the predominant mutation is N370S which accounts for approximately 75 percent of mutant alleles among Jewish patients and approximately 30 percent of the alleles among non-Jewish patients. Homozygosity for N370S is characterized by relatively milder phenotypes (although the phenotype is very heterogenous and severe cases are seen24). N370S has heretofore been considered “protective” against the development of neuronopathic features. The second common mutation found almost exclusively among Ashkenazi Jews is one that usually causes a severe phenotype. Five or six common mutations account for approximately 97 percent of alleles among Jews, but account for less than 75 percent of alleles among non-Jews.8,25,26,27 Although controversial, premarital/prenatal screening for common mutations has become frequent among Ashkenazi Jews.28,29,
The second most common mutation is L444P, which when homozygous accounts for most patients with the neuronopathic type 3 disease, and is the most prevalent mutation in Asians, Arabs, and Norrbottnians. Patients with the unique variant of progressive calcifications of cardiac valves, type 3c, are uniformly homozygous for a point mutation D409H.12
Despite some relationship between specific mutations and the clinical course, genotype–phenotype correlation is imperfect. Elucidation of the three-dimensional structure of the glucocerebrosidase by crystallography has also not improved prediction of disease severity based on the location of mutations in the native protein.30
The majority of the mutations cause glucocerebrosidase misfolding, which may lead to early degradation of the enzyme in the endoplasmic reticulum.31,32 The investigation of the proteotoxic effect of the misfolded mutant enzyme in the endoplasmic reticulum has led to the development of the new therapeutic modality of pharmacologic chaperones (PCs). PCs are targeted to stabilize the mutated glucocerebrosidase and allow its appropriate trafficking from endoplasmic reticulum to Golgi and, finally, to the lysosome.
Three major types of Gaucher disease are differentiated clinically based on absence (type 1) or presence of neurologic features (types 2 and 3).33 Table 72–1 summarizes key clinical, genetic, and demographic features. Although it has been suggested that there is a phenotypic continuum,34,35 it is still useful to think of Gaucher disease as three distinct forms to facilitate genetic counseling and management decisions.
| TYPE 1 | TYPE 2 | TYPE 3 | |||||
|---|---|---|---|---|---|---|---|
| Subtype | Asymptomatic | Symptomatic | Neonatal | Infantile | 3a | 3b | 3c |
| Common genotype | N370S/N370S or 2 mild mutations | N370S/other or 2 mild mutations | Two null or recombinant mutations | One null and one severe mutations | None | L444P/L444P | D409H/D409H |
| Ethnic predilection | Ashkenazi Jews | Ashkenazi Jews | None | None | None | Norrbottnians, Asians, Arabs | Palestinian Arabs, Japanese |
| Common presenting features | None | Hepatosplenomegaly, hypersplenism, bleeding, bone pains | Hydrops fetalis; congenital ichthyosis | SNGP, strabismus, opisthotonus, trismus | SNGP; myoclonic seizures | SNGP; hepatosplenomegaly growth retardation | SNGP; cardiac valves’ calcifications |
| CNS involvement | None | None | Lethal | Severe | SNGP; slowly progressive neurologic deterioration | SNGP; gradual cognitive deterioration | SNGP; brachycephalus |
| Bone involvement | None | Mild to severe (variable) | None | None | Mild | Moderate to severe; kyphosis (gibbus) | Minimal |
| Lung involvement | None | None to (rarely) severe | Severe | Severe | Mild to moderate | Moderate to severe | Minimal |
| Life Expectancy | Normal | Normal/near-normal | Neonatal death | Death before age 3 years | Death during childhood | Death in mid-adulthood | Death in early adulthood |
There is variability in disease severity of all types of Gaucher disease. Type 1 disease may be asymptomatic and be discovered in the course of population surveys of Ashkenazi Jews,28 or incidentally during evaluation of an unrelated hematologic disorder.
Fatigue is a common complaint, usually not invariably related to anemia, but also quite common in nonanemic patients and may be a result of elevated inflammatory cytokines.36
In symptomatic patients, the spleen is typically enlarged,37 whether barely palpable or massively enlarged causing positional symptoms, such as early satiety or abdominal discomfort. Splenic infarction and subcapsular hematoma are uncommon. Hepatomegaly is usually asymptomatic, but it may cause abdominal discomfort and in splenectomized patients or others with very severe disease, liver fibrosis and later cirrhosis,38 with or without portal hypertension, may occur; hepatocellular carcinoma may evolve.39 An increased incidence of nonalcoholic fatty liver disease has been observed.40
Lymphadenopathy has been described,41 including a severe protein-losing form,42 which is a clinical management problem.
Epistaxis, easy bruising, and hemorrhage after surgical or dental procedures and bleeding during labor are common presenting symptoms. These manifestations usually are related to thrombocytopenia as the result of hypersplenism or marrow replacement by Gaucher cells, but platelet dysfunction and decreased levels of coagulation factors have also been described and hence should be assessed prior to surgical procedure or before delivery.43,44,45 Coagulation factor deficiencies may result from liver disease or consumption coagulopathy.
Reduced hemoglobin levels are also primarily a result of hypersplenism and marrow replacement by Gaucher cells, but additional causes include iron deficiency, vitamin B12 deficiency, and autoimmune hemolysis.46,47
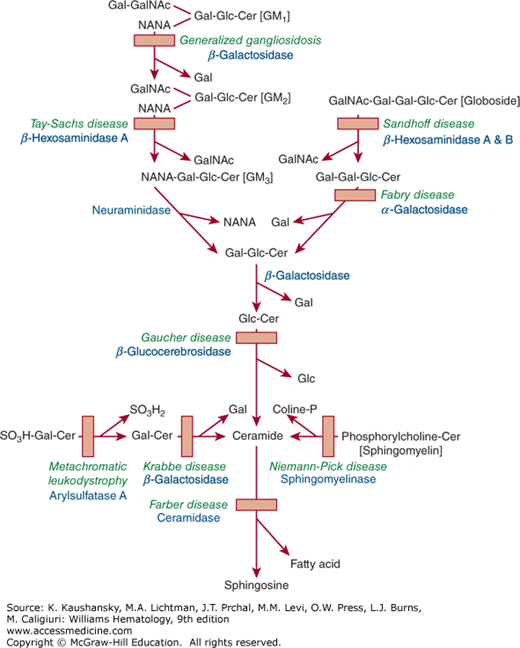
“Gaucheromas” (Fig. 72–2), which are possibly extraosseous in origin48 and/or may mimic a malignant process,49,50 appear idiosyncratically, but possibly after some invasive procedure such as hip surgery; these have been described to be at increased risk of hemorrhaging when manipulated.
Figure 72–2.
A. Histologic section of “Gaucheroma” showing hemorrhagic mass with nucleated red blood cells covered by a fibrous capsule. B. Histologic section at a higher magnification showing nucleated red blood cells admixed with numerous Gaucher cells. (Used with permission ofProf. Eliezer Rosenmann, Shaare Zedek Medical Center, Jerusalem, Israel.)
Severe pulmonary disease with cyanosis and clubbing occurs in some patients with advanced liver involvement, and is usually a consequence of hepatopulmonary syndrome with or without infiltration of the lungs by Gaucher cells.51,52 Mild pulmonary hypertension may be detected by echocardiography,53 but may (rarely) be severe especially among splenectomized patients54; it has not been reported in children.55 Pulmonary function tests may reveal abnormalities, such as reduced diffusion capacity in approximately two-thirds of patients.56
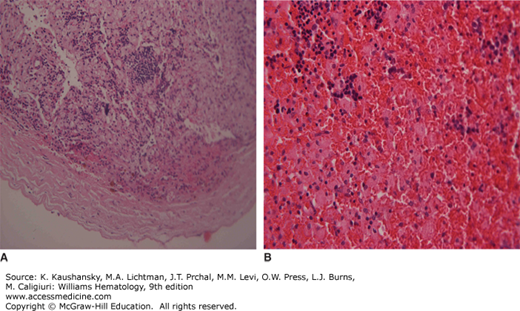
Bone involvement is usually the main cause of morbidity in symptomatic patients and can occur in any long bone.57 Patchy areas of bone demineralization and infarction are seen (Fig. 72–3A), and asymptomatic widening of the distal femur known as Erlenmeyer flask deformity is very common (Fig. 72–3B). Bone metabolism markers indicate that bone resorption predominates,58 but the overall mechanisms underlying development of bone lesions are poorly understood. Children may have delayed bone age and delayed eruption of the teeth.59 Bone pain is probably the most troublesome symptom of Gaucher disease. Bone pain may be related to the pathologic processes evident by radiography, magnetic resonance imaging (MRI), and computerized tomography, or have the character of a “crisis,” which is a self-limiting, albeit exquisitely painful event, associated with signs of acute local and/or systemic inflammation (Fig. 72–3D). Aseptic necrosis of large joints, mainly the femoral heads but also the shoulders and knees (and rarely even in smaller joints) and vertebral collapse are particularly common typically among untreated patients with genotypes resulting in more severe phenotypes (Fig. 72–3C and E).
Figure 72–3.
Gaucher-related skeletal involvement including (A) humerus with chevron or herring-bone pattern; (B) Erlenmeyer flask deformity of the proximal femur; (C) plain radiograph of osteonecrosis of the left hip; (D) magnetic resonance image of pelvis and thighs that was performed 2 weeks after bone crisis of the right thigh. Bone edema is seen in the upper part of the femur at the level of lesser trochanter. Chronic marrow signal changes are seen in both femurs; (E) vertebral collapse. (Used with permission of Dr. Ehud Lebel, Shaare Zedek Medical Center, Jerusalem, Israel.)
Gynecologic and obstetric problems are common and are mainly related to bleeding tendency,60 which may explain why females are more likely to be diagnosed. Delayed menarche and menorrhagia are common, and increased risk of recurrent abortions has been reported.61 Fertility is unaffected in males and females.
Organs other than the spleen, liver, bones, and lungs may be affected. Many patients have pinguecula and a few a pterygium at the corneoscleral limbus.62 Additional findings include uveitis and preretinal white spots in rare cases.63
Renal manifestations are rare and limited to case reports of nephrotic syndrome and renal cell carcinoma.64 Nonetheless, many patients seem to have benign urinary hyperfiltration.65
Neurologic symptoms constitute the hallmark of types 2 and 3 diseases.66 Particularly notable and pathognomonic are oculomotor abnormalities, especially supranuclear gaze palsy (SNGP), which is typically noted horizontally,67 but might occur in the vertical plane as well. Patients with type 2 disease develop hypertonia of the neck muscles with extreme arching of the neck (opisthotonus), bulbar signs, limb rigidity, seizures, and sometimes choreoathetoid movements. In these patients, the SNGP becomes a fixed convergent squint, often facilitating differentiating between type 2 patients, who are terminal by 2 to 3 years of age, and the severe type 3a patients, who may survive longer. Patients with type 3a disease exhibit progressive neurologic abnormalities such as myoclonus and dementia.68 Patients with type 3b disease display aggressive visceral and skeletal involvement but neurologic manifestations are largely limited to horizontal SNGP.68 Patients with type 3c disease exhibit SNGP, mild visceral involvement, and fatally progressive calcifications of mitral and tricuspid valves and of the large arteries.12,68,69,70
Several neurologic abnormalities have been observed in patients with type 1 disease, including peripheral neuropathy71,72 and an increased prevalence of Parkinson disease (the latter also among carriers of a single mutation).73,74,75,76,77 Carriers of severe mutations (e.g., null alleles) were reported to have a 13.6-fold increased risk of Parkinson disease compared to controls, whereas carriers of the more benign mutations have a 2.2-fold increased risk.78 A meta-analysis of patients with Parkinson disease has confirmed this strong association between mutations in the glucocerebrosidase gene and Parkinson disease, which is marked by an earlier age of onset and higher prevalence of cognitive changes.78,79
An increased tendency to infections is sometimes seen, occurring among splenectomized patients or severely affected patients, some of whom may have defective neutrophil chemotaxis.80,81 Bacterial osteomyelitis is most often iatrogenic following surgical intervention at the site of a bone crisis. In children, linear growth retardation is common regardless of disease severity,82 but a compensatory “catch-up” growth may occur by early adulthood.83
There is a higher prevalence of neoplastic disorders in patients with Gaucher disease.84,85 Myeloma has been established to be more prevalent.84,85 Other hematologic malignancies,86 hepatocellular carcinoma, and renal cell carcinoma, may also have increased prevalence.87 Although elevated levels of interleukin-6 in patients with Gaucher disease may link Gaucher disease and myeloma,88 there is no explanation at present for increased incidence of other types of cancer. Some malignancies may be less common.87 The impact of ERT on either an increased or decreased development of malignancies has not been determined.
The complete blood count in patients with Gaucher disease may be normal or may reflect the effects of hypersplenism. A normocytic, normochromic anemia is frequently present, but hemoglobin levels only rarely fall below 8 g/dL. A modest reticulocytosis is often present in anemic patients. The white cell count may be decreased to as low as 1.0 × 109/L, but milder degrees of leukopenia are more common. The differential count is normal, but splenectomized patients tend to show a lymphocytosis. A defect of leukocyte chemotaxis which may be corrected by ERT,89 and in some patients is associated with a tendency to bacterial infections80; monocyte dysfunction has also been reported.81 Thrombocytopenia is typically more prominent than anemia.46 In an anemic patient with an intact spleen and normal range platelet counts, there is probably an alternative reason for the low hemoglobin level, unrelated to Gaucher disease. Thrombocytopenia may be quite severe, even in an otherwise mildly affected patient. In splenectomized patients, anemia is more likely in the absence of thrombocytopenia; white cell count and platelet counts are usually higher than normal. Severe anisocytosis and poikilocytosis also occur in splenectomized patients, with many target cells, some nucleated red cells and Howell-Jolly bodies. During bone crises, leukocytosis, thrombocytosis, and elevated erythrocyte sedimentation are seen. Other markers of inflammation have been noted regardless of disease severity: elevated fibrinogen levels, elevated high-sensitivity C-reactive protein, and increased adhesion and aggregation of red blood cells.90,91
Clotting factor abnormalities may be induced by activated macrophages41,42 or may be found when there is liver involvement. Factor IX deficiency may be a laboratory artifact related to the effect of accumulated lipid on platelet membranes.92 Factor XI deficiency is common among Ashkenazi Jewish patients because of its high coincidental prevalence in this ethnic group.93
Bleeding tendency may also result from defective aggregation or adhesion of platelets33 and therefore platelet function and/or thromboelastography should be tested before surgical and dental procedures and labor.44,94
In most patients, liver function tests are within normal limits but in conjunction with more severe disease, splenectomy, and/or comorbidities (hepatitic B and/or C, or autoimmune diseases) abnormal liver function tests may be seen. Because of the increased prevalence of cholelithiasis,95,96 cholestatic findings may occur. Renal function tests are usually normal.64
Many patients present with polyclonal gammopathies. Monoclonal gammopathies are found in 1 to 20 percent of patients, particularly older patients.79,80,81,82 Increased levels of autoantibodies have been reported,97 and may indicate coincide with autoimmune diseases such as Hashimoto thyroiditis, rheumatoid arthritis, or immune hemolytic anemia.
Biochemical abnormalities have been used as surrogate markers in Gaucher disease. In the past, increased activities of serum acid phosphatase, angiotensin-converting enzyme, serum ferritin, and other hydrolases, such as β-hexosaminidase or β-glucuronidase, were used. Other biomarkers correlate better with the extent of glucocerebroside storage. The most widely used biomarker is chitotriosidase,98 which is undetectable in healthy subjects (its physiologic role is unknown), but is elevated, often several thousand-fold, in patients with Gaucher disease. Chitotriosidase measurement is useful for monitoring both untreated patients, to assess stability versus
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


