INTRODUCTION
SUMMARY
Amyloidosis should be considered in any patient presenting with nephrotic range proteinuria; infiltrative cardiomyopathy or heart failure with preserved ejection fraction; hepatomegaly without specific imaging findings; or peripheral neuropathy, particularly if a monoclonal protein is present; as well as any patient with atypical multiple myeloma.
When a patient is seen with a relevant syndrome, the patient should have immunofixation of serum and urine proteins and measurement of κ and λ immunoglobulin free light chains. If all of these tests are normal, it is unlikely that the patient has immunoglobulin light-chain (AL) amyloidosis.
If any of the above tests are positive, further investigation for amyloidosis should be undertaken. The diagnostic test of choice is subcutaneous fat aspiration; marrow biopsy is the second best procedure. With these two tests, 83 percent of patients will have a positive result when stained with Congo red under green birefringence.
All patients with biopsy proven amyloidosis should have the deposits analyzed by laser capture microdissection mass spectroscopy to definitively classify the exact protein subunit composing the amyloid. This technique does not distinguish between systemic and localized amyloidosis, however.
The prognosis in AL amyloidosis is determined by three tests: (1) the N-terminal probrain natriuretic peptide, (2) serum troponin, and (3) the difference between the involved and uninvolved immunoglobulin free light chains. These three tests can be combined to stage the patient from stage 1 through stage 4.
Treatment of AL amyloidosis involves either standard systemic chemotherapy or high-dose chemotherapy with autologous stem cell transplantation. Fit patients who are expected to have low morbidity with transplantation should undergo this approach. The majority of patients, however, will not be candidates for transplantation and should be treated with traditional systemic chemotherapy; with the cyclophosphamide, bortezomib, and dexamethasone regimen currently favored by many investigators.
Acronyms and Abbreviations
CRAB, calcemia, renal insufficiency, anemia, or bone disease; CyBorD, cyclophosphamide, bortezomib, and dexamethasone; MRI, magnetic resonance imaging; NT-ProBNP, N-terminal probrain natriuretic peptide; TTR, transthyretin.
DEFINITION AND HISTORY
Amyloidosis is a heterogeneous group of diseases characterized by tissue infiltration with misfolded protein precursors as amyloid material. For nearly 100 years, amyloid has been defined by its staining properties. Divry and Florkin first used Congo red to detect amyloid in the brain of patients with Alzheimer disease.1 Amyloid deposits are always extracellular and, under the light microscope, appear amorphous and pink when seen after standard hematoxylin-and-eosin staining. All amyloid deposits bind Congo red dye and demonstrate green birefringence under polarized light,2 and this test remains the standard diagnostic procedure required to confirm a diagnosis of amyloidosis. Under the electron microscope, amyloid consists of rigid, linear, nonbranching fibrils of 9.5 nm diameter.3 Historically, amyloid was defined based on whether there was a family history (inherited), whether it was the result of a chronic inflammatory condition (secondary), or whether it was idiopathic (primary). Today, amyloidosis is classified based on the protein subunit composition of the deposits. The term “primary amyloid” is archaic and should not be used. Instead, such cases should be called “immunoglobulin light-chain (AL) amyloidosis,” reflecting the fact that they represent a systemic plasma cell dyscrasia. The subunit classification is important because AA amyloidosis (secondary) can be the consequence of sustained inflammation but can also be inherited, as in familial Mediterranean fever. Inherited forms of amyloidosis are generally composed of mutant transthyretin (TTR) but can also be a consequence of mutations in apolipoprotein, fibrinogen, and gelsolin.4 Senile systemic amyloidosis, primarily a cardiac disorder, is being increasingly recognized, and is caused by deposition of wild-type TTR as amyloid material in patients older than age 60 years.5 Table 108–1 lists the nomenclature of the various forms of amyloidosis. Practicing hematologists and oncologists are not likely to see patients with forms of amyloidosis other than immunoglobulin light-chain (AL) amyloidosis. This is the only form that is responsive to chemotherapy and is the focus of this chapter; additionally, the term amyloidosis, unless specified otherwise, refers to AL amyloidosis.
| Amyloid Type | Subunit Protein | Clinical Organ Involvement |
|---|---|---|
| AL (κ or λ) or AH | Immunoglobulin light or heavy chain May be localized or systemic | Heart Kidney Liver Nerve |
| AA | Secondary serum amyloid A | Kidney Gastrointestinal Thyroid |
| ATTR (age relelated) | Senile systemic transthyretin | Heart Carpal tunnel |
| ATTR (mutant) | Familial transthyretin | Heart Nerve |
| A Lect-2 | Leukocyte chemotactic factor No mutation found | Kidney |
| A Ins | Insulin localized to injection sites | Skin |
| A Fib | Fibrinogen A-2 mutation | Kidney |
| A β2M | β2-Microglobulin Chronic dialysis | Soft tissue Joints spine |
EPIDEMIOLOGY
Amyloidosis is rare, with an incidence of eight per million persons per year with a median age at diagnosis of 67 years.11 This makes it one-sixth as common as myeloma and twice as common as Waldenström macroglobulinemia. Conversely, a practicing oncologist should see approximately one light-chain amyloid patient for every six patients with myeloma in the oncologist’s practice. If this is not the case, there is a significant likelihood that the disease is going unrecognized.
ETIOLOGY AND PATHOGENESIS
The fibrils of immunoglobulin light chain amyloid are composed, in most patients, of fragments of immunoglobulin light chains. A normal immunoglobulin light chain has a molecular weight of approximately 25 kDa, but the immunoglobulin light chains found in amyloid deposits usually range from 8 kDa to 15 kDa. The fragment size is important because it usually reflects the deletion of the constant region of the immunoglobulin light chain, making immunohistochemistry a poor technique to identify the fibril type in paraffin-embedded tissues.6 A fraction of patients have immunoglobulin heavy chain amyloid deposits where fragments of immunoglobulin G, A, or M heavy chains comprise the basic subunit. Whereas AL amyloidosis represents approximately 62 percent of all amyloid patients, immunoglobulin heavy-chain amyloidosis represents less than 1 percent of all patients.7
There is clearly something “amyloidogenic” about the immunoglobulin light chains in patients with AL amyloidosis. Unlike monoclonal gammopathy and myeloma where the κ:λ ratio is 2:1, in AL amyloidosis, the κ:λ ratio is 1:3, suggesting an intrinsic propensity of λ light chains to form amyloid. Moreover, the subgroup λVI is exclusively found in patients with AL amyloidosis.8 When the light chains are extracted from the urine of patients with myeloma and injected into mice, amyloid deposits are not seen. However, when the light chains from patients with amyloidosis are extracted from the urine and injected into mice, they will develop amyloidosis.8 The exact structural characteristics that lead to the misfolding of the α helical protein into the amyloid β-pleated sheet configuration are unknown. Patients with amyloidosis are classified into those with myeloma and those without myeloma. The percentage of plasma cells present in the marrow at the time of diagnosis is prognostic and predicts outcome in patients with amyloidosis.9 As a result, the percentage of plasma cells in the marrow may dictate alternate therapeutic considerations at diagnosis (see “Therapy” below). Overt myeloma with CRAB criteria (hypercalcemia, renal insufficiency, anemia, or bone disease) is uncommon in AL amyloidosis. However, an elevation in the percentage of plasma cells in the marrow of patients without CRAB criteria confers the same adverse prognosis as observed in patients who have overt symptomatic myeloma, and these groups can be considered as a single cohort of myeloma-associated amyloidosis. Patients who do not have myeloma at presentation have almost no chance of developing myeloma later in the course of their disease.10 The plasma cells in the marrow of patients with amyloidosis tend to be nonproliferative and frequently lack the karyotypic abnormalities typically seen in myeloma, such as –17p, t(4;14), and –13. A translocation 11;14 is commonly seen in amyloidosis and appears to confer an inferior outcome. Circulating plasma cells detectable by flow cytometry are uncommonly seen relative to their frequency in myeloma12
CLINICAL FEATURES
Unfortunately, the symptoms of amyloidosis are vague and often nonspecific. Physical findings, when present, can be highly specific but are only present in a minority of patients. For a disease this rare, it is important for clinicians to have an operational approach to avoid inappropriate resource utilization. Only 1 percent of patients with AL amyloidosis are younger than age 40 years. Consequently, it would be unlikely for younger patients to have AL amyloidosis, although this age group regularly can be seen with secondary systemic amyloidosis. The majority of patients with AL amyloidosis are males (66 percent), unlike myeloma, where the male-to-female is 52:48.15
The most common symptoms reported by patients with amyloidosis are fatigue, weight loss, and lower-extremity edema. Unfortunately, these symptoms are too nonspecific to be used as a trigger to screen patients, as the yield for these vague symptoms would be very low. The mechanism underlying fatigue in this disorder is usually amyloid heart disease, which can be an extremely subtle diagnosis and is discussed further under the specific amyloid syndromes. Weight loss accompanying amyloidosis can be impressive, and may exceed 20 kg. This usually results in an investigation for occult malignancy; but even if blind biopsies are performed, the diagnosis may be overlooked if a specific request for amyloid stains is not made to the pathologist. Lower-extremity edema may be attributable to nephrotic range proteinuria, hypoalbuminemia, and transudation of serum into the extracellular space. Edema may also be seen from high filling pressures caused by the restrictive cardiomyopathy, which, again, can easily be overlooked because of the subtle diagnostic testing required. These patients are often treated empirically with diuretics until cardiac dysfunction becomes more evident.
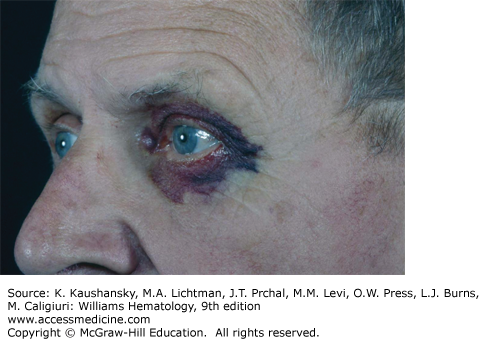
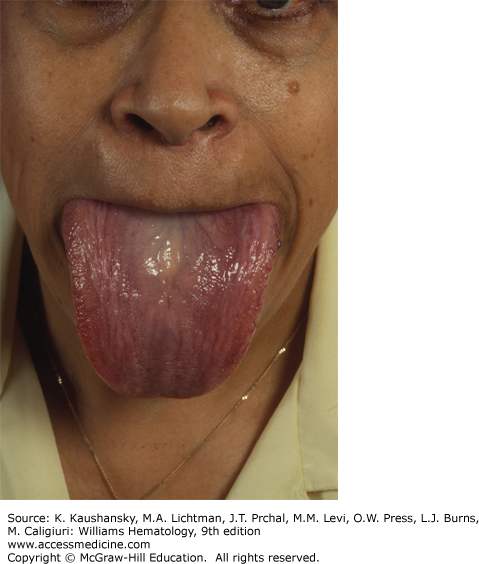
The physical findings of amyloidosis are present in only one patient in six. Periorbital purpura is frequently cited as a diagnostic finding and, when present, is useful but is only seen in 15 percent of patients (Fig. 108–1). Sometimes, the purpura is subtle and can only be seen with the patient’s eyes closed. Even when purpura is present, it may lead to an evaluation of a coagulopathy or platelet dysfunction, which would invariably be negative in this setting. Purpura not only is seen on the eyelids but also on the face, neck, and anterior chest above the nipple line. Enlargement of the tongue is seen in approximately one person in eight (Fig. 108–2). Dental indentations on the underside of the tongue are characteristic. Often the patient will have sudden-onset of sleep apnea syndrome as a result of occlusion of the airway by the enlarged tongue when the patient is supine.16 Even when diagnostic findings are present, their significance is often missed, leading in many cases to tongue biopsy with a preoperative suspicion of tongue cancer. Most patients with enlargement of the tongue will also have palpable submandibular lymphadenopathy, usually caused by displacement of the nodes by the enlarged base of the tongue; although amyloid infiltration of the submandibular gland may also result in a firm palpable mass in the submandibular region. Hepatomegaly is seen in approximately 20 percent of patients and may be due to direct infiltration of the liver, which will cause a firm markedly enlarged liver.17 In some patients, however, the liver enlargement is a reflection of high venous filling pressures in the right-sided cardiac chambers and represents chronic passive congestion. Patients rarely have periarticular infiltration of the shoulders producing the so-called shoulder pad sign, a baseball-shaped enlargement of the anterior soft tissues of the shoulder. A rare patient will develop temporal artery infiltration and develop classic jaw claudication, as well as limb, buttock, and calf claudication.18 On questioning, many patients will have xerostomia from infiltration of the minor salivary glands; and at some centers, biopsy of the minor salivary glands is a preferred technique for the diagnosis of amyloidosis.19
AMYLOID SYNDROMES
Because the symptoms of amyloidosis are highly nonspecific and the physical findings are specific but not very sensitive, an operational approach is required to ascertain which patients need investigation for amyloidosis. We recommend screening for AL amyloidosis when a patient is seen with any of the following clinical syndromes:
Nephrotic range proteinuria with any serum creatinine level
Infiltrative cardiomyopathy or heart failure with preserved ejection fraction. A normal ejection fraction does not exclude AL amyloidosis
Hepatomegaly or alkaline phosphatase elevation without specific imaging abnormalities
A mixed axonal demyelinating peripheral sensory, motor or autonomic neuropathy, particularly when associated with a monoclonal gammopathy
A patient with myeloma with symptoms that are not typical of the disease, particularly profound, unexplained fatigue
Table 108–2 gives the frequency of amyloid syndromes seen in patients at the Mayo Clinic. If an oncologist sees a patient with one of these five syndromes, or an internist is faced with an undiagnosed patient, laboratory evaluation is indicated, as outlined below.
The best screening tests for initial evaluation of patients with suspected amyloidosis are immunoelectrophoresis and immunofixation of both serum and urine and a serum immunoglobulin free light-chain assay (both κ and λ). Systemic immunoglobulin AL amyloidosis is a plasma cell dyscrasia, and 99 percent of patients will have a detectable abnormality of one of these three tests, reflecting synthesis by a clonal population of plasma cells in the marrow. If an immunoglobulin protein is detected, further investigation for amyloidosis as described in the next section (“Differential Diagnosis”) should proceed. If a systemic plasma cell dyscrasia and an immunoglobulin light chain cannot be confirmed, three possibilities exist: (1) the patient does not have amyloidosis, (2) the patient does not have systemic amyloidosis, or (3) the amyloidosis is not immunoglobulin light chain in type and reflects a different protein subunit.
The serum free light-chain assay is a critically important test. It not only heightens the suspicion of the presence of immunoglobulin AL amyloidosis, it also is prognostic and vital to staging the patient. The serum immunoglobulin free light chain is part of the response evaluation for this disease. A screening serum protein electrophoresis is insufficient as a screening technique because a visible M-spike is seen in less than half of patients because of the high prevalence of primary light-chain proteinemia.
Finding a monoclonal protein in the serum or in the urine of a patient with heavy albuminuria often obviates the need for a renal biopsy. A patient with free light chains in the serum or urine and proteinuria can have only one of three disorders: (1) myeloma cast nephropathy, (2) AL amyloidosis, or (3) Randall-type immunoglobulin deposition disease (κ).
Screening for a light-chain immunoglobulin is the best noninvasive approach when confronted with a patient with any of the five syndromes listed in Table 108–2. If amyloid is present but the light chains are normal, strong consideration of referral to a specialty center to further clarify the underlying form of amyloidosis should be considered.
Once a clinician has begun an evaluation of a patient with a compatible clinical syndrome and an immunoglobulin abnormality has been detected, a biopsy is required to confirm the diagnosis before therapy should commence. Although imaging of amyloid deposits with various radionuclides, most notably anti–serum amyloid P component, continue to be explored and used in practice, they are not a substitute for histologic confirmation of the presence of amyloidosis.20,21,22 In a patient with renal, cardiac, hepatic, or peripheral nerve amyloidosis, it is straight forward to establish the diagnosis by kidney, heart, liver, or nerve biopsy, but this is unnecessary in the majority of patients. Subcutaneous fat aspiration is a bedside office procedure done with local anesthesia and does not require an incision (Fig. 108–3). An excellent YouTube video has been produced by the Boston Medical Center on the technique (https://www.youtube.com/watch?v=tctYTmxd9gQ). Typically, fat aspirations are reviewed at a specialty center unless the pathology department regularly processes fatty tissue. The fat is not paraffin-embedded and, therefore, it is subject to overfixation, trapping of Congo red dye, and the possibility of inadequate controls.23 Fat aspiration is positive in three-quarters of patients with amyloidosis.
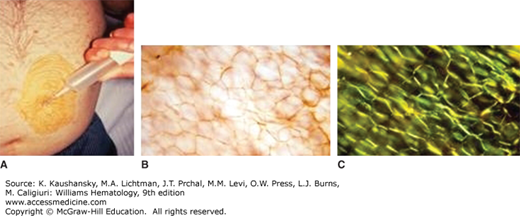
A second tissue easily stained for amyloid is the marrow. Because AL amyloidosis patients will have a plasma cell dyscrasia, a marrow examination is clinically indicated to quantify the number of plasma cells in the marrow. Congo red staining of marrow blood vessels can detect amyloid deposits in half of patients with AL amyloidosis. Combining the two techniques (marrow and fat pad aspiration), establishes a diagnosis in 85 percent of afflicted patients. Minor salivary gland biopsy, endoscopic gastric biopsy, rectal biopsy, and skin biopsy can also be used to establish the diagnosis. If the clinical suspicion of AL amyloidosis is strong and these biopsies are negative, it is appropriate to biopsy the affected organ.
Once tissue containing Congo red has been identified, it becomes imperative that the protein subunit be determined. Historically, immunohistochemistry has been used to identify the type of amyloid, but immunohistochemistry can be challenging because only those protein subunits for which antisera exist can be detected.24,25 Second, particularly in AL amyloidosis, the reactive epitope may have been deleted during the process of amyloid fibril formation and, frequently, the background staining is high, particularly in kidney tissues. Third, because the protein in amyloid misfolds, even if the epitopes are present, they may be hidden deep within the deposits and may be inaccessible to commercial antisera. Not only can immunohistochemistry be unreliable, it can be misleading. It is therefore recommended that laser capture of the amyloid deposit be performed routinely followed by mass spectroscopic analysis.26,27,28
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


