INTRODUCTION
SUMMARY
The antiphospholipid (aPL) syndrome (APS) is an acquired thrombophilic disorder in which patients have vascular thrombosis and/or pregnancy complications attributable to placental insufficiency, accompanied by laboratory evidence for the presence of antiphospholipid antibodies in blood. The disorder is referred to as primary APS when it occurs in the absence of systemic lupus erythematosus (SLE), and secondary APS in its presence. Any portion of the circulatory tree can be affected, although the most frequently affected vessels are the deep veins of the lower extremities. Abnormalities that have been reported in association with the syndrome include virtually all other autoimmune disorders, immune thrombocytopenia, acquired platelet function abnormalities, hypoprothrombinemia, acquired inhibitors of coagulation factors, livedo reticularis, heart valve abnormalities, atherosclerosis, pulmonary hypertension, and migraine. Rare patients have a catastrophic form of APS (CAPS) in which there is disseminated thrombosis in large- and small-vessel thrombi, often after a triggering event such as infection or surgery, and often with multiorgan ischemia and infarction.
APS is a misnomer, the main antigenic targets for thrombogenic aPL antibodies are epitopes on phospholipid-binding proteins, the most important of which appears to be β2-glycoprotein I (β2GPI). The syndrome is identified by persistent abnormalities of laboratory tests for antibodies against these phospholipid–protein cofactor complexes, detected by immunoassays and by coagulation assays (also known as “lupus anticoagulant assays”) that, paradoxically, report the inhibition of phospholipid-dependent coagulation reactions. Long-term warfarin anticoagulant therapy is the usual treatment for thrombosis in patients with APS, although there is some controversy about whether treatment of patients with APS stroke might be better treated with aspirin. Patients with recurrent spontaneous pregnancy losses and APS generally are treated with aspirin and heparin for prophylaxis against deep vein thrombosis during their pregnancies and the postpartum period. CAPS patients have a high mortality and, in addition to anticoagulants, often require plasmapheresis and immunosuppressive agents. Patients without clinical manifestations of APS or a history of SLE should generally not undergo diagnostic screening for aPL antibodies and, if tested and found to be positive, should not be committed to antithrombotic therapy solely on the basis of laboratory abnormalities.
Acronyms and Abbreviations
aCL, anticardiolipin; APC, activated protein C; aPL, antiphospholipid; APS, antiphospholipid syndrome; aPTT, activated partial thromboplastin time; ARDS, acute respiratory distress syndrome; ASIA, autoimmune/autoinflammatory syndrome induced by adjuvants; AVWS, acquired von Willebrand syndrome; BFP syphilis test, biologic false-positive serologic test for syphilis; β2GPI, β2-glycoprotein I; CAPS, catastrophic APS; CMV, cytomegalovirus; dRVVT, dilute Russell viper venom time; ELISA, enzyme-linked immunosorbent assay; HCQ, hydroxychloroquine; Ig, immunoglobulin; IL, interleukin; LA, lupus anticoagulant; LDL, low-density lipoprotein; LMWH, low-molecular-weight heparin; MAPK, mitogen-activated protein kinase; NOACs, new oral anticoagulants; RVV, Russell viper venom; SCR, short consensus repeat; SLE, systemic lupus erythematosus; TLR, toll-like receptor; TM, thrombomodulin; t-PA, tissue-type plasminogen activator; UFH, unfractionated heparin; VWF, von Willebrand factor.
DEFINITION AND HISTORY
The antiphospholipid (aPL) antibody syndrome (APS) is a disorder in which vascular thrombosis or pregnancy complications attributable to placental insufficiency occur in patients with laboratory evidence for antibodies directed against proteins that bind to phospholipids. The syndrome was first proposed to be a distinct entity, “the anticardiolipin (aCL) syndrome,” in 19851 and soon was renamed APS.2 While precise data are not available, the syndrome is thought to affect approximately 10 percent of patients with venous thrombosis,3,4 and approximately 20 percent of women with three unexplained fetal losses before 12 weeks of gestation, or at least one intrauterine fetal death after 12 weeks of gestation.5
The term “aPL antibodies” can refer to (1) antibodies that recognize protein-phospholipid complexes as in cofactor-dependent aCL assays, (2) antibodies that recognize the proteins directly as in anti-β2glycoprotein I assays (anti-β2GPI), (3) an abnormal coagulation test in several assays that report inhibition of phospholipid-dependent coagulation reactions, collectively as termed lupus anticoagulant (LA) tests, and (4) antibodies that recognize phospholipid directly as in syphilis and are not associated with the APS disease entity.
A brief review of the history of APS6,7,8 helps explain the confusing terminology (Table 131–1); the reader is referred to references 6,7,8 for more detailed accounts. In retrospect, the first assay for aPL autoantibodies was Moore and Mohr’s report of the “biologic false-positive” serologic tests for syphilis (BFP syphilis test) in 19529; this abnormality came to be associated with systemic lupus erythematosus (SLE).10 The contemporaneous introduction, of the activated partial thromboplastin time (aPTT), which used cephalin, a phospholipid extract of animal brains, as the “partial thromboplastin” (distinct from the “complete thromboplastin,” tissue factor, and phospholipid),11 led to the recognition of a unique type of anticoagulant in patients with SLE, that was frequently associated with BFP syphilis tests.12 Because the of its initial association with SLE this phenomenon was misnamed LA.13 It became recognized that the LA was purely an in vitro phenomenon that was not limited to SLE and that was not associated with bleeding complications unless another hemostatic defect was present.6 Furthermore, the LA came to be associated with recurrent pregnancy losses14,15 and with thrombotic and embolic manifestations.16 The development, in 1983, of the aCL antibody assay, that measured antibodies against the anionic phospholipid, cardiolipin (diphosphatidylglycerol), the primary antigen in the syphilis test reagent,17 was the advance that led to the identification of a new syndrome. Within a few years, it became recognized that aPL antibodies did not bind phospholipids directly but instead were directed against proteins that bound to the phospholipid, primarily β2GPI (see “Pathogenesis” below). This information became important in helping to unravel the mechanisms for APS and in advancing diagnostic testing toward the goal of distinguishing between the syndrome and incidental false-positive tests. Table 131–2 describes the current consensus investigational criteria for diagnosing APS.18
| Immunoassay Path | Coagulation Path |
|---|---|
| 1950s: Syphilis testing | 1950s: Partial thromboplastin time inhibitor |
| 1970s: Lupus anticoagulant (LA) | |
| 1980s: Antiphospholipid (aPL) antibody enzyme-linked immunosorbent assay (ELISA; e.g., anticardiolipin [aCL] immunoassays) | 1980s: Recognition that LAs are inhibitors of phospholipid-dependent coagulation reactions |
| 1990s: Anticofactor ELISA (anti-β2-glycoprotein I [β2GPI], antiprothrombin, etc.) | |
| 2005: Demonstration that antibodies against domain I of β2GPI are associated with increased risk of thrombosis | 2004: Demonstration that resistance to the anticoagulant effect of annexin A5 correlates with thrombosis in antiphospholipid syndrome |
Clinical
|
Laboratory
|
Most patients with elevated aPL antibodies do not have the syndrome; elevated aPL antibody levels can occur in patients with several types of infections that induce formation of antibodies recognizing anionic phospholipids directly, patients taking medications such as chlorpromazine or procainamide, and even in normal healthy individuals. Testing of patients who have neither clinical manifestations of the disorder or SLE for aPL antibodies should be discouraged because it incurs the risk of inappropriate diagnostic and treatment decisions.
ETIOLOGY AND PATHOGENESIS
As with most autoimmune conditions, the etiology of APS is not understood. It has been demonstrated that even normal healthy individuals have memory B cells that produce aPL antibodies. In a study of patients with infectious mononucleosis, 10 to 60 percent of immunoglobulin (Ig) M aPL-producing cells expressed CD27, the marker of memory B cells.19 The affinity of aPL antibodies for their target becomes increased by the inclusion of amino acids lysine, arginine, and asparagine within the complementary determining regions of the heavy and light chains.20
Although antibodies against anionic phospholipid moieties arise during the course of infections such as syphilis and Lyme disease, those are distinct from antibodies generated by patients with the syndrome because they generally recognize phospholipid epitopes directly (also referred to as “cofactor independent”) and are not associated with the clinical manifestations of the syndrome. There are intriguing hints for molecular mimicry mechanisms and that infection and vaccination-induced APS could be related to autoimmune/autoinflammatory syndrome induced by adjuvants (ASIA).21 aPL antibodies have been reported in patients who developed thrombosis after varicella infection,8,22,23 and in patients with hepatitis C.24,25 aPL antibodies were reported in a patient with cytomegalovirus (CMV) infection, mesenteric and femoropopliteal thrombosis.26,27 β2GPI cofactor-dependent antibodies against cardiolipin, phosphatidyl serine, and phosphatidyl ethanolamine have been identified in sera from patients with parvovirus B19.28 Bacterial infections are a predisposing risk factor for the catastrophic form of APS (CAPS).29 A high proportion of HIV-1 patients have aPL antibodies; more than 40 percent in one study, in which 18 percent had aCL and 30 percent had anti-β2GPI (mostly of the IgA isotype),30; however, positivity for these antibodies was not associated with thrombosis. A link has been proposed between the cardiac valvular disease in acute rheumatic fever and the presence of aPL antibodies.31 aCL antibodies having β2GPI dependence and LA activity have been generated in rabbits immunized with lipid A and lipoteichoic acid, suggesting that some bacteria can contribute to the production of pathogenic aPL antibodies.32 It has also been proposed that cellular apoptosis, with the resulting exposure of anionic phospholipids on cell surfaces, may trigger the generation of aPL antibodies.33,34,35 Molecular mimicry between β2GPI-related synthetic peptides and structures within bacteria, viruses, and tetanus toxoid36 have been demonstrated in an experimental model for APS.37 Mice immunized with a CMV-derived peptide developed aPL antibodies and thrombosis, and showed evidence for endothelial cell activation.38
Reports of familial clustering of raised aPL antibody levels39 indicate that genetic susceptibility can play a role in their development. In one study of 84 APS patients, more than 35 percent had at least one relative, and more than 20 percent had two or more relatives, with evidence of at least one clinical feature of APS, such as thrombosis or recurrent fetal loss.40
It has been clearly established in a number of experimental animal models for APS that aPL antibodies play a causal role in the development of thrombosis and pregnancy loss.41,42,43,44,45 Although it is reasonable to assume that the same holds for the human disorder, the epitopic specificities of the autoantibodies that cause disease and the mechanisms by which they produce clinical manifestations require further elucidation.
Antibodies against phospholipid that arise during the immunologic response to syphilis and other infections (with the notable exception of leprosy46) recognize anionic phospholipid epitopes directly,47 whereas pathogenic aPL antibodies recognize phospholipid-binding proteins, primarily β2GPI.48,49
β2GPI (also named apolipoprotein H), a member of the complement control protein or short consensus repeat superfamily,50 is a highly glycosylated single-chain plasma protein composed of 326 amino acids, with a molecular weight of approximately 50 kDa (Fig. 131–1). β2GPI has five short consensus repeat (SCR) stretches of approximately 60 amino acids45 (also referred to as complement control protein [CCP] repeats). Epitopic specificities for individual domains may have pathogenic and prognostic significance (see “Immunoassays” below).51,52,53,54
Figure 131–1.
Schematic of the conformational states of β2GPI. The unbound protein is in a closed conformation in which the epitope on domain I (DI) is shielded by a portion of domain V (DV). Binding to phospholipid membranes via a “barb,” consisting of a hydrophobic loop formed by Ser311 to Lys317, near the carboxyterminus of DV, requires the protein to open and exposes an immunogenic epitope near the aminoterminal portion of the molecule. (Reproduced with permission from Rand JH A snappy new concept for APS. Blood 2010 Aug 26;116(8):1193–1194.)
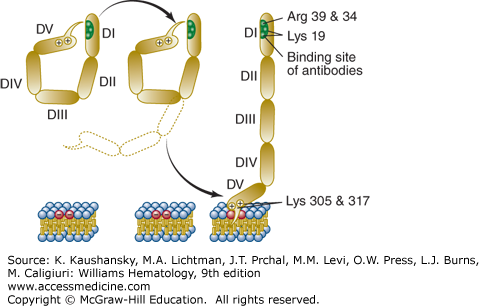
The affinity of β2GPI for anionic phospholipids derives from cationic residues from its aminoterminus that have affinity for anionic polar heads of phospholipids and a hydrophobic loop which inserts into the lipid bilayer. β2GPI has five domains for which antibodies have been identified. IgG antibodies against an epitope comprising Gly40-Arg43 in the domain I of β2GPI have been reported to have a stronger correlation with thrombosis than antibodies against other epitopes.51 Recent data support the concept that β2GPI undergoes conformational changes that may be important for the APS disease process. By transmission electron microscopy, unbound β2GPI appears to be in a closed conformation because of the affinity of a portion of carboxyterminal domain V for the protein’s amninoterminal domain I, where the phospholipid binding site is located near the carboxy-terminus of SCR domain V (see Fig. 131–1).55 The binding of β2GPI to anionic phospholipid membranes, requires a conformational change which exposes an epitope in domain I that had been cryptic in the unbound conformation (see Fig. 131–1).55
Although β2GPI bind to phospholipids, its role in aPL-mediated cell signaling (described in the section Proposed Pathogenic Mechanisms), is mediated via binding to toll-like receptors (TLRs) and not by direct binding to the lipid bilayer.
While the in vivo biologic function(s) of the protein has (have) not been defined, several interesting properties have been demonstrated. The molecule binds to apoptotic cells,56 and may play a role in their phagocytosis and clearance.57 β2GPI binds to oxidized low-density lipoprotein (LDL) and may play a role in its clearance.58 β2GPI binds to lipopolysaccharide and the scavenged complex is taken up by monocytes/macrophages.59 β2GPI reduces platelet adhesion to collagen in flow chambers by interfering with the platelet–von Willebrand factor (VWF) interaction by binding to its A2 domain, thereby interfering with its binding to the platelet glycoprotein Ib complex.60 β2GPI may also promote fibrinolysis as a cofactor for tissue-type plasminogen activator (t-PA) via its SCR domain V, which increases fibrinolytic activity.61 The protein may have a further effect on fibrinolysis by binding to endothelial cells via annexin A2, a protein that also serves as a receptor for plasminogen and t-PA.62 Homozygous β2GPI-null mice have not been demonstrated to display a thrombotic phenotype.63 However, the protein may play a role—though not a critical one—in the reproductive process, as there was a reduction in the number of viable implantation sites in β2GPI-null mice and reduced fetal weight and fetal-to-placental weight ratio in late gestation, suggesting compromised placental function.64
In addition to β2GPI, a number of other antigenic targets have been identified for aPL, including, but not limited to, prothrombin, coagulation factor V, protein C, protein S, annexin A2, annexin A5, high- and low-molecular-weight kininogens, and factors VII/VIIa and vimentin–cardiolipin complex.65,66,67,68
Table 131–3 and Fig. 131–2 summarize several of the main current hypotheses for pathogenic mechanisms in APS. The mechanisms of the human APS disease process have been difficult to elucidation, mainly for two reasons: (1) The phenotypes of vascular thrombosis and pregnancy morbidity are not unique to APS, so it is difficult to ascertain whether the candidate mechanism is playing a causal role or is incidental. (2) Antibodies isolated from APS patients recognize a multiplicity of antigenic determinants69,70 that can have a broad range of effects, so it is difficult to determine which specificities and effects are responsible for disease manifestations in humans.
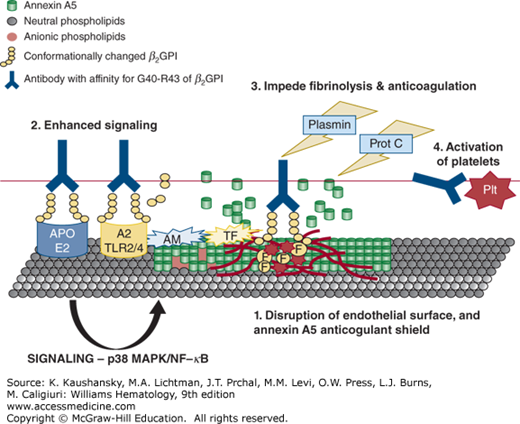
Figure 131–2.
Multiple pathogenic mechanisms of aPL antibodies. (1) On a disrupted endothelial surface, anti-β2GPI–β2GPI complexes bind through the cationic domain V of β2GPI to anionic structures, such as heparan sulfate to provide a prothrombotic surface. (2) In addition, anti-β2GPI–β2GPI complexes activate endothelial cell receptors such as ApoE2, TLR2/TLR4 and annexin A2 to promote downstream signaling pathways involving p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor-κB (NF-κB), leading to the upregulation of tissue factor (TF) and adhesion molecules (AM) and a proinflammatory/prothrombotic phenotype. (3) Anti-β2GPI–β2GPI complexes also impede fibrinolysis and anticoagulation by impeding plasmin, annexin A5 anticoagulant activity, and the protein C (Prot C) pathways. (4) Anti-β2GPI–β2GPI complexes bind to directly to activate platelets (Plt) and promote aggregation.
|
Disruption of the Endothelial Surface and Annexin A5 Anticoagulant Shield Annexin A5 is a potent anticoagulant protein with high affinity for phospholipid membranes that contain anionic phospholipids, specifically phosphatidyl serine.71 Annexin A5 forms two-dimensional crystals over the phospholipid bilayers that shield them from binding coagulation factors72 and it has been proposed that the protein may play a thrombomodulatory role on the surfaces of cells lining the placental and systemic vasculatures. Annexin A5 is highly expressed on the apical membranes of placental syncytiotrophoblasts, the location where maternal blood interfaces with fetal cells.73 Pregnant annexin A5-null mice develop placental infarctions of fetuses and yield reduced litter sizes.74 Pregnant mice treated with anti–annexin A5 antibodies developed placental necrosis, fibrosis, and pregnancy loss.75 Dissociation of annexin A5 from the surface of human placental trophoblasts and human umbilical vein endothelial cells accelerates the coagulation of plasma exposed to those cells.76 Annexin A5 binds to the surfaces of endothelial cells and inhibits thrombin formation.77
aPL antibody–antigen complexes disrupt the crystallization of annexin A5 and displace the protein from phospholipid membrane surfaces (Fig. 131–3).78,79,80,81 In contrast to the LA phenomenon, aPL antibodies accelerate coagulation reactions in systems that include annexin A5.78,82,83,84,85 IgG fractions from APS patients reduce the quantity of annexin A5 on cultured placental trophoblasts76,86 and endothelial cells76,87 and accelerate the coagulation of plasma exposed to these cells.76 This effect of aPL antibodies on annexin A5 binding has been correlated with IgG antibodies that recognize a specific epitope—domain I of β2GPI in patients with APS who have thrombosis52 and spontaneous pregnancy losses. Figure 131–2 includes a model for this mechanism.
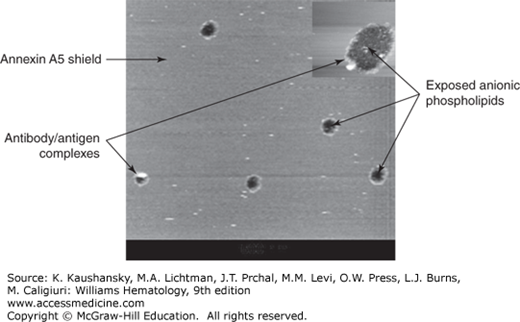
Figure 131–3.
Disruption of annexin A5 shield by monoclonal antiphospholipid antibodies and β2-glycoprotein I (β2GPI). Atomic force microscopy picture showing the effect of a monoclonal aPL antibody on a preformed annexin A5 crystal. The figure demonstrates the smooth lipid bilayer covered by the annexin A5 crystals, disrupted by antibody–β2GPI complexes (white circles) and exposing anionic phospholipids (black holes) to coagulation factors and accelerated coagulation. (Modifed with permission from Rand JH, Wu XX, Quinn AS, et al: Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay. Am J Pathol 163(3):1193–1200, 2003.)
Binding to Endothelial Surface Receptors Enhances Cell Signaling aPL antibodies can bind, injure, and activate cultured vascular endothelial cells.88,89,90,91 Cultured endothelial cells incubated with aPL antibodies with specificity for cell surface β2GPI express increased levels of cell adhesion molecules92 triggered by their binding to cell surface β2GPI.93 Annexin A2 serves as a receptor for β2GPI,62 and anti-β2GPI antibodies may thereby stimulate expression of tissue factor on endothelial cells.94 In animal models, the signaling effects of aPL antibodies were significantly reduced in mice treated with an anti-annexin A2 monoclonal antibody and also in annexin A2-null transgenic mice.95 These effects of the aPL antibodies may be mediated by TLR-4 of the innate immunity system,96,97 although there are data indicating participation of other TLRs, particularly TLR-2.98 This binding results in downstream signaling that involves TRAF6 (tumor necrosis factor receptor-associated factor 6) and MyD88 (myeloid differentiation factor 88).99 Increased expression of tissue factor is mediated by p38 mitogen-activated protein kinase (MAPK).100
Binding of autoantibodies to annexin A2 may also promote thrombosis by inhibiting fibrinolysis. APS patients have increased titers of antibodies against annexin A2, an endothelial surface receptor for t-PA, and plasminogen.101 The blocking of annexin A2 by aPL antibodies impedes plasmin generation in a t-PA–dependent generation assay, and inhibits cell surface plasmin generation on human umbilical vein endothelial cells.94 Several additional mechanisms have been identified by which aPL antibodies can interfere with fibrinolysis. β2GPI is a cofactor for t-PA–mediated activation of plasminogen, and aPL antibodies against β2GPI interfere with its binding to t-PA, thereby downregulating plasminogen activation.61 Finally, fibrinolysis may also be impaired by autoantibodies directed against the catalytic site of plasmin or t-PA,102,103 by an increased level of plasminogen activator inhibitor-1,104 and by inhibition of autoactivation of factor XII with ensuing reductions of kallikrein and urokinase.105
Apolipoprotein E receptor 2 (apoER2), a member of the LDL-receptor family is found on endothelial surfaces,106 monocytes,107 and platelets, and may also serve as receptor for anti-β2GPI–β2GPI complexes where it can also trigger the phosphatidylinositol 3′-kinase (PI3K)/Akt pathway108 and increase tissue factor and cell adhesion molecule expression. IgG-mediated dimerization of β2GPI and binding to ApoER2′ increases the sensitivity of platelets to agonists of aggregation.109
Complement-Mediated Injury Complement activation may play a role in the APS disease process. The IgG2 subtype of aPL most closely correlates with thrombosis.110,111 Blockade of complement activation using a C3 convertase inhibitor or genetic deletion of C3 protected mice from pregnancy complications induced by aPL antibodies.112,113,114 These effects involve the aPL-stimulated expression of tissue factor by myeloid cells,115 and to involve proteinase-activated G-protein–coupled receptor (PAR)-2 signaling,116 indicating that complement activation can be pathogenic via both direct injury and downstream signaling.
Induction of Tissue Factor Activity in Leukocytes aPL antibodies can promote tissue factor expression by leukocytes.115,117,118,119 The specific binding site on these cells has not been elucidated.
Inhibition of Fibrinolysis and Endogenous Anticoagulation aPL antibodies can interfere with fibrinolysis in several ways. Antibodies against annexin A2, an endothelial surface receptor for t-PA and plasminogen, can interfere with binding of plasminogen and t-PA and thereby reduce plasmin formation and fibrinolysis.94,101,103 Monoclonal aPL antibodies derived from APS patients can directly inhibit plasmin’s enzymatic activity.102 β2GPI is a cofactor for t-PA–mediated activation of plasminogen,120 and anti-β2GPI can interfere with this activity. Also, it has been reported that women with APS have significantly increased levels of circulating plasminogen activator inhibitor-1 (PAI-1), implying impaired fibrinolysis.104
Interference with Components of the Protein C Activation Pathway The protein C pathway (Chaps. 114 and 139) is initiated by thrombin binding to thrombomodulin (TM), which activates protein C bound to the endothelial protein C receptor (EPCR). Activated protein C (APC), together with free protein S, then proteolyses coagulation factors Va and VIIIa. APC also modulates signaling events by interfering with PAR-1 signalling.121,122 aPL antibodies can interfere with the activation of protein C by TM–thrombin and with the activity of APC, as well as protect factors Va and VIIIa from proteolysis by APC.65 Acquired APC resistance has been described in APS plasmas123 and has been correlated with anti-β2GPI domain I antibodies,124 a risk factor for thrombosis. The presence of antibodies against EPCR in APS patients was proposed to be a risk factor for fetal death.125
Antiphospholipid Antibodies Activate Platelets An experimental animal model that includes in vivo imaging has provided data indicating that aPL-induced thrombosis is a consequence of platelet activation that then promotes endothelial activation and fibrin formation.126 aPL antibodies can induce platelet aggregation,127 an effect that might be promoted via signaling through apoER2 receptors; the β2GPI binding site for apoER2 on platelets was localized to its domain V.128 As described above (see “Antigenic Specificities”), β2GPI also has a dampening effect on platelet adhesion by interfering with the platelet–VWF interaction, and consequently aPL antibodies, by interfering with this β2GPI-mediated dampening, can increase platelet adhesion in flow systems.60
Other Mechanisms APS patients have been shown to have autoantibodies against tissue factor pathway inhibitor.129 Some aPL antibodies cross-react with heparin and heparinoid molecules, which are highly polyanionic, and hence, inhibit their contribution to antithrombin activity.69 aPL antibodies show cross-reactivity against oxidized LDL130 and are associated with an increased risk of atherosclerosis.131 Antibodies against β2GPI-oxidized LDL complexes have been proposed to be atherogenic by reducing their clearance.132 Finally, in addition to promoting thrombosis, aPL antibodies may contribute to other vascular lesions by stimulating the mammalian target of rapamycin complex (mTORC) pathway (Fig. 131–4).133
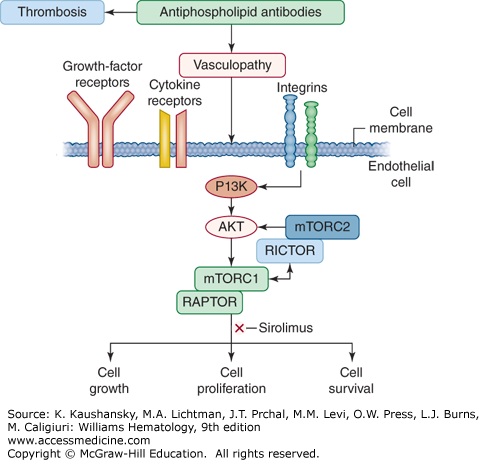
Figure 131–4.
Pathogenesis of vasculopathy in the antiphospholipid syndrome. In addition to promoting thrombosis, antiphospholipid antibodies also trigger vasculopathy by binding to vascular endothelial cells and activating the mammalian target of rapamycin (mTOR) signaling pathway. Extracellular and intracellular signals through the phosphatidylinositide 3′-kinase (PI3K)-AKT pathway activate mTOR pathway which regulates cell growth, proliferation and survival. The mTOR enzyme is a component of two complexes, mammalian target of rapamycin complex (mTORC) 1 and mTORC2. The activity of mTORC1 is regulated by a subunit of the regulatory-associated protein of mTORC1 (RAPTOR), whereas the activity of mTORC2 is regulated by a subunit of the rapamycin-insensitive companion of mTOR (RICTOR). (Modifed with permission from Eikelbloom JW, Weitz JI: The mTORC pathway in the antiphospholipid syndrome. N Engl J Med 371(4):369–371, 2014.)
CLINICAL FEATURES
Table 131–4 summarizes the clinical features of APS. Patients generally present with thrombotic manifestations, that is, evidence for vasoocclusion or end-organ ischemia or infarction, and/or pregnancy losses and complications attributable to placental insufficiency. The usual age at presentation with thrombosis is approximately 35 to 45 years. Except for patients with SLE, men and women are equally susceptible to thrombotic manifestations. No differences have been observed between the arterial and venous distributions of thromboses of primary and secondary APS patients.134
|
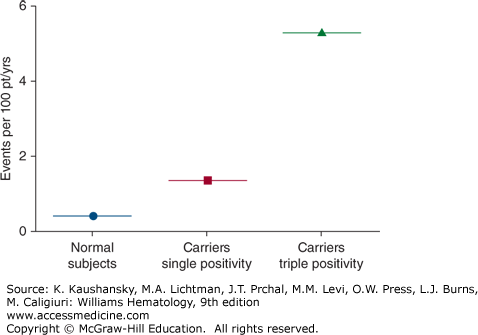
Patients can present with spontaneous venous and/or arterial thrombosis or embolism in any site; however, about half of all patients have deep vein thrombosis of the lower extremities.135,136 Other sites of venous thromboembolic events include pulmonary embolism, thoracic veins (superior vena cava, subclavian vein, or jugular vein), and abdominal or pelvic veins.136 Approximately one-fourth of patients present with arterial thromboses; the remainder present with concurrent arterial and venous thrombosis.136 Patients may also present with stroke, cerebral venous thrombosis, upper-extremity venous thrombosis,135 myocardial infarction, adrenal infarction, acalculous gallbladder infarction, aortic thrombosis with renal infarction,120 and mesenteric artery thrombosis.137,138 Thrombosis may occur spontaneously or in the presence of some other risk factor such as estrogen replacement therapy, oral contraceptives,134,139 vascular stasis, surgery, or trauma. Women are at particularly high risk for venous thrombosis during pregnancy and in the postpartum period.134 Some APS patients with venous thrombosis have concurrent genetic thrombophilic conditions such as the factor V Leiden variant, and it has been postulated that this may increase the risk of thrombosis.140,141,142,143 Concurrent positivity for all three aPL antibody assays—that is, the aCL antibody assay, anti-β2GPI antibody assay, and the LA assay—appears to be a highly significant risk factor for having an initial thrombotic event (Fig. 131–5).144 If this finding is confirmed, it could identify a group of patients who might warrant consideration for prophylactic treatment. A history of having had a thromboembolic event is probably the most significant risk factor for recurrence of venous thromboembolism in APS, with a reported frequency that reached approximately 30 percent in patients followed for 4 years after the first episode.145 The risk of recurrent thromboembolism correlates with the titer of antibodies,145,146 and with the presence of LA. In addition, it appears that the presence of anti-β2GPI domain I antibodies significantly increase the risk of thrombosis, compared to patients who have antibodies that are not domain I dependent and LA that is not domain I dependent.51
Figure 131–5.
Average annual rates of first cardiovascular events (including venous thromboembolism) among antiphospholipid (aPL) antibody–negative and aPL antibody-positive populations. Concurrent positivity for all three aPL antibody assays—that is, “triple positivity” for the aCL antibody assay, anti-β2GPI antibody assay, and the lupus anticoagulant assay—is a highly significant risk factor for a first thrombotic event. (Modified with permission from Pengo V, Ruffatti A, Legnani C et al: Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: A multicenter prospective study. Blood 118(17):4714–4718, 2011.)
APS patients frequently present with other autoimmune conditions. A significant proportion of SLE patients have elevated aPL antibodies, with estimates ranging between 12 and 30 percent for aCL antibodies and 15 and 34 percent for LA antibodies.147 APS has been associated with the concurrence of other autoimmune conditions including, but not limited to, rheumatoid arthritis,148 Sjögren syndrome,149 myasthenia gravis,150 Budd-Chiari syndrome in the setting of SLE,151 Graves disease,152 autoimmune hemolytic anemia, progressive systemic sclerosis,153 Evans syndrome,154 Takayasu arteritis,155 polyarteritis nodosa,156 and immune thrombocytopenia (see section Thrombocytopenia below and also Chap. 117).
The most common neurologic manifestations of APS are stroke or transient ischemic attacks (TIAs), which were the initial presentation of up to 30 percent of adults with APS in a large European cohort.157 In the European Catastrophic Antiphospholipid Antibody Syndrome (CAPS) Registry, cerebral manifestations occurred in 62 percent of patients and caused 13 percent of deaths.158 Recurrent strokes are more likely in patients with APS and other risk factors for cerebrovascular disease, such as cigarette smoking, hypertension hyperlipidemia, oral contraceptive use, and SLE.159,160
Most APS patients with stroke have arterial thromboembolic occlusive events that are clinically indistinguishable from the more common arteriosclerotic strokes. APS should be suspected in young patients with TIAs or stroke, particularly when the more typical risk factors for cerebrovascular disease are absent.161 Cerebral venous thrombosis is less common in APS patients and presents at a younger age, and is more extensive than in non-APS patients with the disorder.162 In one series of 40 cases of cerebral venous thrombosis, three patients (8 percent) had elevated aPL antibody levels.163 Superior sagittal sinus thrombosis has been reported with primary APS.164
There is controversy about whether migraines in patients with aPL antibodies should be regarded as thromboocclusive events.165 Other neurologic abnormalities reported to be associated with aPL antibodies include seizures/epilepsy,166 chorea, Guillain-Barré syndrome, transient global amnesia, dementia, diabetic peripheral neuropathy, and orthostatic hypotension.167 Recurrent acute transverse myelopathy has been described with APS.168,169,170,171,172 However, in one study of 315 SLE patients, including 10 with a history of transverse myelopathy, that disorder was not associated with aPL antibodies.173 Multiple sclerosis patients have a high incidence of elevated aCL antibody levels (in one series, 9 percent had IgG antibodies and 44 percent had IgM antibodies),174 however, no clinical distinctions were found between aPL-positive and aPL-negative patients and the antibodies do not appear to be associated with thrombosis. Patients with psychotic disorders have an increased prevalence of LA and aCL antibodies, even in the absence of treatment with antipsychotic drugs.175
CAPS is a relatively infrequent but devastating presentation of APS, is characterized by severe widespread vascular occlusions.176 Diagnostic criteria for CAPS include evidence of involvement of at least three organs, systems, and/or tissues; development of manifestations simultaneously or in less than 1 week; histopathologic confirmation of small-vessel occlusion; and laboratory confirmation of the presence of aPL antibodies.177 According to the CAPS Registry, a web-based database of 433 patients with CAPS (https://ontocrf.costaisa.com/en/web/caps/), the majority of CAPS patients are female (69 percent), in their late thirties (mean age of 38.5 years), but the condition can present at any age (range: 0 to 85 years). In half of the CAPS cases, the patients’ catastrophic event was their first APS manifestation. Precipitating factors of CAPS include infections, drugs (sulfur-containing diuretics, captopril, and oral contraceptives), surgical procedures, and cessation of prior anticoagulant therapy. In 26.9 percent of cases, the patients also had SLE. The most frequently affected organ was the kidney (73 percent of episodes), followed by lungs (58.9 percent), the brain (55.9 percent), the heart (49.7 percent), and the skin (45.4 percent). Other organs were also affected, including the peripheral vessels, intestines, spleen, adrenal glands, pancreas, retina, and marrow. Patients present with evidence for severe multiorgan ischemia/infarction, often with concurrent disseminated microvascular thrombosis. Patients with CAPS can present with renal insufficiency, respiratory failure resulting from acute respiratory distress syndrome (ARDS) and pulmonary emboli, cerebral manifestations such as encephalopathy, stroke and seizures, cardiac problems such as heart failure, myocardial infarction and valvular defects, and skin complications such as livedo reticularis and skin necrosis.178 Most patients show histologic evidence of microangiopathy mainly affecting small vessels of the kidneys, lungs, brain, heart, and liver. Only a minority of patients experience large-vessel occlusions. Laboratory evidence for disseminated intravascular coagulation is frequently present. An LA is present in 81.7 percent of patients, and the aCL IgG is the most common positive aPL antibody.178 Improved treatment has reduced mortality from approximately 50 percent to approximately 20 percent.176 Relapse is rare in survivors. The only identified predictive factor for adverse outcome is underlying SLE.179
At this time, aPL screening of otherwise asymptomatic with no history of prior complications obstetrical patients is not warranted because of the high frequency of false-positive tests; most studies have estimated the prevalence of aPL antibodies among general obstetric populations to be approximately 5 percent or less and most of these aPL-positive patients are not clinically affected.180
Among obstetric patients with recurrent fetal losses, approximately 16 to 38 percent have aPL antibodies. In addition, pregnant women with elevated aPL antibodies had significantly more obstetric complications, including preeclampsia, abruption placentae, miscarriage, prematurity, intrauterine fetal demise, intrauterine growth restriction, and oligohydraminos, than aPL antibody-negative pregnant women.181,182,183 In approximately half of patients, the pregnancy losses occur in the first trimester. Other patients present with later losses, most in the second trimester, but some even later, including stillbirth. Pregnancy complications attributable to APS include three or more recurrent spontaneous first trimester miscarriages, one or more fetal losses during the second trimester, stillbirth, episode of preeclampsia, preterm labor, placental abruption, intrauterine growth restriction, and oligohydramnion.184,185,186 Pregnant patients with APS are also more prone to developing deep vein thrombosis during pregnancy or the puerperium. Rarely, pregnant patients develop CAPS.187,188 The best predictor for pregnancy loss in a patient who tests positive for aPL antibodies has been demonstrated to simply be a previous history of pregnancy loss, complications, or thrombosis.146,189 A recent study reported that any aPL antibody-positive women with an unexplained early loss prior to 10 weeks have a higher risk of complications in their second pregnancy190 but the degree of laboratory abnormalities were not associated with increased risk. Positivity by more than one assay appears to correlate with increased pregnancy morbidities.182,191 Histologic abnormalities were found in many, but not all, placentas of aPL patients.192 Studies of placental pathology in patients with aPL antibodies, but without a prior history of fetal loss, showed that approximately half had evidence of uteroplacental vascular pathology, approximately half had evidence of thrombotic occlusion, and approximately one-third had chronic villitis and/or decidual plasma cell infiltrates.193,194
Overall, it appears that the presence of aPL antibodies does not affect the success rate of implantation. Although one group has reported data that indicated that women with recurrent implantation failure were more likely to have positive assays for aPL antibodies compared to fertile negative controls,195 a review of 29 studies showed mixed results.196 Many of the studies were noted to have limitations, including problems with study design and statistical power. The current consensus is that aPL antibodies are not a cause of infertility.197 Recently, the14th International Congress on Antiphospholipid Antibodies Task Force also concluded that “there are no data to support the inclusion of infertility as criteria for APS and investigation of APS in patients with infertility should not be done in routine clinical practice, being reserved only for research purposes.”196
The cutaneous manifestations of APS may comprise the first signs of APS in some patients.198,199 Livedo reticularis is relatively common, occurring in 24 percent of a series of 1000 aPL patients,200 and occasionally presents in a necrosing form.201 Noninflammatory vascular thrombosis is the most frequent histopathologic feature. Necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, cutaneous ulceration and necrosis, erythematous macules, purpura, ecchymoses, painful skin nodules, and subungual splinter hemorrhages, anetoderma (macular atrophy), discoid lupus erythematosus, and cutaneous T-cell lymphoma have all been reported.
aPL antibodies are associated with increased susceptibility to coronary artery disease,202 particularly premature atherosclerosis.203,204 APS should be considered in patients who lack the usual risk factors for coronary artery disease and in patients with evidence for thrombotic or embolic coronary artery occlusion that lack angiographic evidence of atherosclerotic disease. aPL antibodies appear to be a risk factor for adverse outcomes following all coronary revascularization procedures,202 and for restenosis after percutaneous transluminal coronary angioplasty.205,206 An ultrasound study of carotid arteries provided evidence supporting an association of aPL antibodies with premature atherosclerosis; relatively young primary APS patients (mean age: 37 ± 11 years) had significantly increased intimal medial thickness compared to control non-APS groups.207
Approximately 35 percent of patients with primary APS have cardiac valvular abnormalities detected by echocardiography.208
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


