INTRODUCTION
SUMMARY
Thrombotic microangiopathy is a general term for the combination of microangiopathic hemolytic anemia and thrombocytopenia, often accompanied by signs and symptoms consistent with disseminated microvascular thrombosis. Thrombotic thrombocytopenic purpura (TTP) refers to thrombotic microangiopathy, without an obvious predisposing condition, and without oliguric renal failure. TTP is caused by autoantibodies to ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif member 13), a plasma metalloprotease that normally cleaves von Willebrand factor (VWF) and regulates VWF-dependent platelet aggregation. Inherited deficiency of ADAMTS13 causes congenital TTP, which typically responds to plasma infusion. Most patients with acquired TTP respond to plasma exchange, although many have relapsing disease. Hemolytic uremic syndrome (HUS) refers to thrombotic microangiopathy that usually causes oliguric or anuric renal failure. Ingestion of Shiga toxin-producing Escherichia coli can cause the most common or “typical” form of HUS that is usually preceded by bloody diarrhea. Inherited or acquired defects in the regulation of the alternative complement pathway cause HUS referred to as “atypical” because it occurs without a prodrome of bloody diarrhea. Secondary thrombotic microangiopathy can occur in association with metastatic cancer, infections, organ transplantation, and certain drugs. These variants of thrombotic microangiopathy differ in pathogenesis and prognosis, but can be difficult to distinguish because their clinical features often overlap.
Acronyms and Abbreviations
ADAMTS, a disintegrin and metalloprotease with thrombospondin repeats; aHUS, atypical hemolytic uremic syndrome; ANA, anti-nuclear antibody; APS, antiphospholipid syndrome; aPTT, activated partial thromboplastin time; CFH, complement factor H; CFHR, complement factor H-related protein; DDAVP, desmopressin; DGKE, diacylglycerol kinase ε; Gb3, globotriaosylceramide 3; HELLP, hemolysis, elevated liver enzymes, low platelet count; HIT, heparin-induced thrombocytopenia; HUS, hemolytic uremic syndrome; LDH, lactate dehydrogenase; MCP, membrane cofactor protein; MMACHC, methylmalonic aciduria and homocystinuria type C protein; PT, prothrombin time; SLE, systemic lupus erythematosus; STEC, Shiga toxin-producing Escherichia coli; Stx, Shiga toxin; TM, thrombomodulin (gene name THBD); TTP, thrombotic thrombocytopenic purpura; VEGF, vascular endothelial growth factor; VWF, von Willebrand factor.
THROMBOTIC THROMBOCYTOPENIC PURPURA
Thrombotic thrombocytopenic purpura (TTP) refers to thrombotic microangiopathy without another apparent cause and without acute renal failure, although mild or modest renal insufficiency may be seen. Tissue injury can affect almost any organ but often results in neurologic damage. TTP is associated with autoantibodies against the plasma metalloprotease ADAMTS13 (a member of the “a disintegrin and metalloprotease with thrombospondin repeats” family) that reduce plasma ADAMTS13 activity to less than 10 percent of normal.
Eli Moschcowitz reported the first detailed description of TTP in 1924.1 The patient was a 16-year-old girl with fever, severe anemia, leukocytosis, petechiae, and hemiparesis. Her renal function was not impaired, but the urine contained albumin, hyaline casts, and granular casts. She became comatose and died 2 weeks after her first symptoms. At autopsy, hyaline thrombi were found diffusely in terminal arterioles and capillaries, particularly of the heart and kidney. For many years, patients with similar findings were said to have Moschcowitz disease. The name TTP was proposed in 19472 and widely adopted thereafter.
In 1966, a review of 272 published cases defined the major clinical features of TTP.3 Most patients were females between the ages of 10 and 39 years. The symptoms and physical findings included thrombocytopenia, hemolytic anemia with numerous fragmented red cells or schistocytes, neurologic findings, renal damage, and fever. Mortality exceeded 90 percent; the average hospital stay was only 14 days before death, and 80 percent of patients lived fewer than 90 days after the onset of symptoms. However, dramatic recoveries occurred in some cases following splenectomy.
This grim prognosis was recorded before a report in 1976 that whole blood exchange transfusions induced prompt remissions in eight of 14 patients.4 Similar responses were described after plasmapheresis with plasma replacement.5 One remarkable case report showed that plasmapheresis was effective if the replacement fluid was plasma or cryoprecipitate-depleted plasma, but ineffective if the replacement fluid contained just albumin.6 Furthermore, simple plasma infusions without plasmapheresis could induce sustained remissions, suggesting that replacement of a missing plasma factor sometimes was sufficient to ameliorate TTP.6
These reports led to the widespread adoption of plasma therapy for TTP, and two studies published in 1991 provided compelling evidence for its efficacy. Plasma infusion was associated with 91 percent survival in 108 patients, an impressive improvement over historical experience.7 The same year, a prospective randomized comparison of plasma exchange and plasma infusion in 102 patients with TTP was reported.8 Long-term survival was 78 percent for the plasma exchange group and 63 percent for the plasma infusion group, a significant difference in favor of plasma exchange.
A link between TTP and von Willebrand factor (VWF) was proposed in 1982, based on studies of four patients with chronic relapsing TTP.9 Their plasma VWF multimers were much larger than those of healthy controls and similar in size to the VWF multimers secreted by endothelial cells. Patients with TTP were proposed to lack a depolymerase activity, perhaps a protease or a reductase, that shortens newly secreted VWF multimers in vivo and produces the multimer distribution of normal plasma. The absence of this depolymerase would cause the persistence of “unusually large” VWF, which promotes intravascular platelet aggregation, thrombocytopenia, and microvascular thrombosis. Plasma exchange therapy could provide the missing depolymerase activity, or remove other factors that provoke clinical relapses.
A candidate depolymerase was identified in 1996, when a metalloprotease in plasma was shown to cleave VWF multimers subjected to high fluid shear stress or to mild protein denaturants.10,11 Soon thereafter, children with congenital TTP were shown to have inherited deficiency of this metalloprotease,12 and adults with acquired TTP were shown to have autoantibody inhibitors of the enzyme.13,14 The VWF cleaving protease was purified,15,16 cloned,17,18 and named ADAMTS13, a new member of the ADAMTS family of metalloproteases. Simultaneously, the ADAMTS13 locus was identified by linkage analysis in families affected by congenital TTP and causative ADAMTS13 mutations were characterized.19
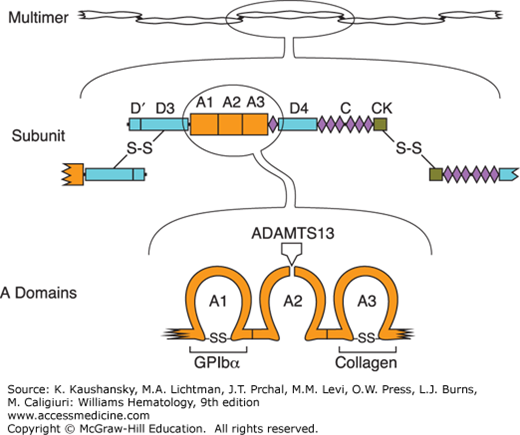
TTP is caused by unregulated VWF-dependent platelet thrombosis. Large VWF multimers mediate platelet adhesion at sites of vascular injury by binding to connective tissue and to glycoprotein Ib (GPIb) on the platelet surface (Chap. 120). The VWF subunit from which multimers are constructed has a modular structure consisting of five types of conserved structural motifs (Fig. 132–1). VWF multimers bind to collagen through domain A3, and to platelet GPIb through domain A1. When platelets bind to VWF under conditions of high fluid shear stress, the VWF multimer is stretched and the Tyr1605-Met1606 bond within domain A2 becomes accessible to ADAMTS13, which cleaves it and thereby can release any adherent platelets. ADAMTS13 deficiency prevents this feedback inhibition of platelet adhesion and leads to widespread microvascular thrombosis. ADAMTS13 levels greater than 10 percent appear sufficient to prevent thrombotic microangiopathy.
Figure 132–1.
Structure of von Willebrand factor (VWF). Multimeric VWF (top) is composed of identical subunits with four kinds of structural motifs, including three A domains, six C domains and a homologous D4N domain, two complete and one partial D domains, and a cystine knot (CK) domain. Subunits (middle) are linked into multimers by disulfide bonds between C-terminal CK domains and N-terminal D3 domains. Domain A1 (bottom) binds platelet glycoprotein Ibα (GPIbα), domain A3 binds collagen in extracellular matrix, and domain A2 contains a Tyr-Met bond that is susceptible to cleavage by ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif member 13).
ADAMTS13 deficiency in TTP is caused by polyclonal autoantibodies against ADAMTS13, usually immunoglobulin (Ig) G but occasionally IgA or IgM.13,14 These antibodies almost always bind the ADAMTS13 spacer domain, and often bind to the CUB domains and first thrombospondin-1 repeat; they bind less frequently to other thrombospondin-1 repeats, the metalloprotease domain, or the propeptide.20,21,22 Most patients have autoantibodies that inhibit ADAMTS13 activity. The rest have noninhibitory antibodies that are likely to mediate clearance of ADAMTS13 from the circulation.23
The annual incidence of TTP reportedly is two to six per million population in the United States and approximately 2.2 per million in the United Kingdom.24,25 Seasonal or geographical trends have not been observed consistently. The demographics of TTP are similar to those of systemic lupus erythematosus (SLE). TTP is relatively uncommon before age 20 years, with a peak incidence between ages 30 and 50 years.24,25 Across many reports the female-to-male ratio averages approximately 2:1, but female preponderance is more pronounced below age 50 years and the ratio approaches equality above age 60 years.24,25 Other risk factors for TTP include African ancestry26,27 and obesity.27,28 Women have a tendency to present during late pregnancy or peripartum (reviewed in Refs. 29 and 30). HLA-DRB1*11 is overrepresented severalfold in whites with TTP.31,32
The onset of TTP can be dramatically acute or insidious, developing over weeks. Approximately one-third of patients have symptoms of hemolytic anemia.3,29 Thrombocytopenia typically causes petechiae or purpura; oral, gastrointestinal or genitourinary bleeding is less common but can be severe. Many patients describe an antecedent upper respiratory tract infection or flu-like illness. Abdominal pain and tenderness are common. Nausea, vomiting, and diarrhea may occur, but bloody diarrhea is uncommon.
Systemic microvascular thrombosis typically affect the kidney, heart, brain, pancreas, adrenals, skin, spleen, marrow, and most other tissues except the lungs, which are spared. Renal involvement is common, but acute renal failure occurs in fewer than 10 percent of cases.26,27,29 Neurologic findings can be transient or persistent and may include headache, visual disturbances, vertigo, personality change, confusion, lethargy, syncope, coma, seizures, aphasia, hemiparesis, and other focal sensory or motor deficits.3,29 The frequency of neurologic findings or fever has decreased from more than 90 percent to approximately 50 percent over the past 40 years,3,8,26,27,29 probably because these features no longer are recognized as necessary to diagnose TTP.
The symptoms of TTP sometimes can be quite atypical, either at first presentation or upon relapse. Thrombocytopenia without hemolytic anemia may herald the onset of disease. In rare instances, visual disturbances, pancreatitis, stroke, or other thrombosis may precede overt thrombotic microangiopathy by days to months.33,34,35,36 Macrovascular venous or arterial thrombosis occurs in up to one-half of patients.37
Cardiac involvement may cause chest pain, myocardial infarction, congestive heart failure or arrhythmias.29,38,39 Direct pulmonary involvement is uncommon but severe acute respiratory distress syndrome may occur, possibly secondary to cardiac failure.29 Gastrointestinal symptoms are common and can include abdominal pain, nausea, vomiting, and diarrhea.3,29 Physical examination may suggest acute pancreatitis or mesenteric ischemia. Infrequent findings include Raynaud phenomenon, arthralgia, myalgia, and retinal hemorrhage or detachment.3,29
The symptoms and signs of TTP are nonspecific. The diagnosis depends on laboratory testing to document microangiopathic hemolytic anemia and thrombocytopenia, without another predisposing cause. Anemia is almost universal with a mean hemoglobin of approximately 8 g/dL.27,40 Thrombocytopenia typically is severe, with a mean platelet count of approximately 20 × 109/L.26,27,40 Hemolysis is indicated by an elevated reticulocyte count and serum lactate dehydrogenase (LDH), undetectable serum haptoglobin, and increased total and unconjugated bilirubin. Coombs test is almost always negative.7,8 Renal microvascular injury is common with microhematuria, granular or red cell casts, and proteinuria, but the serum creatinine is often normal and seldom greater than 2 mg/dL.7,8,27,40 Approximately 50 percent of patients have a positive anti-nuclear antibody (ANA) test.40
Almost all patients have normal values for plasma fibrinogen, prothrombin time (PT) and activated partial thromboplastin time (aPTT),7,8 reflecting a minor role of blood coagulation in TTP. Evidence of myocardial damage is common, with elevated troponin levels.38,39
The characteristic morphological feature of TTP on the blood film is a marked increase in schistocytes. Schistocytes are helmet cells, or small irregular triangular or crescent shaped cells with pointed projections, that lack central pallor (Chap. 2).41 Patients with TTP often have markedly increased schistocytes; in a study of six patients, schistocytes comprised a mean of 8.3 percent of all red cells with a range of 1 percent to 18.4 percent.42 Spherocytes also may be seen.
ADAMTS13 activity is characteristically less than 10 percent and this degree of acquired ADAMTS13 deficiency appears to be specific for TTP.13,14,43,44 If adult patients with thrombotic microangiopathy are selected with no plausible secondary cause, no diarrheal prodrome, and no features suggestive of hemolytic uremic syndrome (HUS) (e.g., oliguria, severe hypertension, dialysis, serum creatinine >3.5 mg/dL), then at least 80 percent of those selected have ADAMTS13 activity less than 10 percent of normal. The majority of patients with severe ADAMTS13 deficiency have autoantibody inhibitors13,14,26,45 and almost all patients have autoantibodies that bind ADAMTS13 by enzyme-linked immunosorbent assay (ELISA).
Depending on the clinical context, laboratory tests should be considered to detect conditions that may cause thrombotic microangiopathy by mechanisms other than ADAMTS13 deficiency such as pregnancy, cobalamin deficiency, SLE and other autoimmune diseases, antiphospholipid syndrome (APS), HIV, and Shiga toxin–producing organisms.
The histologic appearance of microvascular lesions in TTP is consistent with a pathophysiologic role of VWF-dependent platelet thrombosis. Amorphous thrombi and subendothelial hyaline deposits may be found in the small arterioles and capillaries of any organ, but are particularly common (in order of decreasing severity) in the myocardium, pancreas, kidney, adrenal gland and brain. The liver and lung are relatively spared. The lesions consist mainly of platelets and VWF, with little fibrin and few inflammatory cells. They often include focal endothelial cell proliferation.46,47
The diagnosis of TTP should be entertained for any patient with microangiopathic hemolytic anemia and thrombocytopenia, without evidence for disseminated intravascular coagulation, and without features associated with Shiga toxin–producing Escherichia coli (STEC)-HUS such as a prodromal diarrheal illness and acute oliguric or anuric renal failure. These criteria can only be approximate, however, because many diseases associated with secondary thrombotic microangiopathy can produce overlapping clinical and laboratory findings. As a consequence, making a diagnosis of TTP can be a challenge and a wide differential diagnosis often must be considered (Table 132–1).
Thrombotic Thrombocytopenic Purpura (TTP) Autoimmune, with antibodies against ADAMTS13 |
Congenital Thrombotic Thrombocytopenic Purpura (Upshaw-Schulman Syndrome) Inherited ADAMTS13 deficiency, with mutations in ADAMTS13 |
| Shiga Toxin-Producing Escherichia Coli Hemolytic Uremic Syndrome (STEC-HUS) |
Atypical Hemolytic Uremic Syndrome (aHUS) Alternative complement pathway defects Diacylglycerol kinase ε (DGKE) defects |
Secondary Thrombotic Microangiopathy Disseminated intravascular coagulation Infections (viral, bacterial, fungal) Streptococcus pneumoniae Tissue transplant-associated Chemotherapy or radiation injury Tissue rejection Graft-versus-host disease Cancer Pregnancy associated (preeclampsia, eclampsia, HELLP [hemolysis, elevated liver enzymes, low platelet count] syndrome) Autoimmune disorders Systemic lupus erythematosus and other vasculitides Antiphospholipid syndrome Drugs (commonly implicated) Immune (quinine, ticlopidine) Toxic (cyclosporine, tacrolimus, mitomycin C, gemcitabine) Cobalamin metabolic defects Malignant hypertension Mechanical hemolysis (e.g., malfunctioning aortic or mitral valve prosthesis) |
Schistocytes occur in a variety of conditions besides TTP, although the level seldom enters the 1 to 18 percent range typical of TTP. For example, schistocytes were seen in the blood film of 58 percent of healthy controls, with a mean of 0.05 percent and a range of 0 to 0.27 percent of all red cells.42 Up to 0.6 percent schistocytes were observed in patients with chronic renal failure, preeclampsia, or properly functioning prosthetic heart valves.42 Severe hemolysis and marked schistocytosis occur in patients with defective mechanical heart valves. Patients receiving marrow allografts or autografts for a variety of indications had a mean of 0.7 percent schistocytes 6 weeks after transplantation, with a range of 0 to approximately 4 percent schistocytes.48,49 Approximately 10 percent of patients had at least 1.3 percent schistocytes, placing them at risk for a diagnosis of thrombotic microangiopathy.49
ADAMTS13 levels are normal to moderately decreased in newborns, during pregnancy, after surgery, and in chronic liver cirrhosis, chronic renal insufficiency, acute inflammatory states, and a variety of thrombocytopenic disorders other than TTP.44,50 Severe sepsis may sometimes cause acquired severe ADAMTS13 deficiency, although the incidence and clinical significance of the finding remain uncertain.51 Some patients with acute viral hepatitis, severe liver cirrhosis52 or venoocclusive disease after stem cell transplantation53 have had severe ADAMTS13 deficiency (<10 percent) at least transiently, which is consistent with the synthesis of ADAMTS13 in liver.17,18,19
The mainstay of therapy for TTP is plasma exchange, which removes antibody inhibitors of ADAMTS13 and replenishes the enzyme. After diagnosing TTP, or determining that the diagnosis is sufficiently likely to justify treatment, plasma exchange therapy should be started as soon as feasible. Studies establishing the value of plasma therapy have excluded secondary thrombotic microangiopathy,7,8 so the efficacy of plasma exchange has been demonstrated directly only for TTP. The optimal dose of plasma is not known, but a common practice is to perform plasma exchange once daily at a volume of 40 or 60 mL/kg, equivalent to 1.0 or 1.5 plasma volumes. For refractory disease, the intensity of plasma exchange can be increased to 1 plasma volume twice daily.54 Prompt treatment is important and if plasma exchange must be delayed more than a few hours, plasma should be given by simple infusion at 20 to 40 mL/kg total dose per day, consistent with the patient’s ability to tolerate the fluid load.55
The replacement fluid should contain ADAMTS13. Satisfactory results have been obtained with fresh-frozen plasma,7,8 plasma cryosupernatant,56,57,58 and various pathogen-inactivated plasma products.59 The incidence of allergic reactions and transfusion-associated lung injury may be lower with solvent/detergent-treated plasma than with fresh-frozen plasma,60 but the incidence of thrombosis may be increased with some preparations.59,61 Cryosupernatant is depleted in the largest VWF multimers but has normal ADAMTS13 levels,62 which could make cryosupernatant particularly suitable for the treatment of TTP. Nevertheless, small randomized trials suggest that cryosupernatant is not superior to fresh-frozen plasma for the initial treatment of TTP.56,57 Methylene blue-treated plasma may be less effective than fresh-frozen plasma,59,63 despite having a similar concentration of ADAMTS13.59
Plasma exchange should be continued daily until the patient has a treatment response as shown by a platelet count greater than 150 × 109/L for at least 2 days.55 Whether plasma exchange then should be simply stopped or tapered is not known. A typical strategy is to reduce the frequency of plasma exchange to every other day (or twice per week) for several days. If the disease remains quiescent, then treatment can be stopped and the patient monitored closely for recurrence. Alternatively, plasma exchange can be stopped abruptly with monitoring for recurrent thrombocytopenia over several days.
TTP is an autoimmune disease and the use of glucocorticoids is logical, although a beneficial effect has not been demonstrated conclusively. Common practice is to give prednisone or equivalent at a total daily dose of 1 mg/kg, in one or two doses, for the duration of plasma exchange, followed by tapering. An alternative regimen is methylprednisolone 1 g intravenously daily for 3 days.55
The use of antiplatelet agents in TTP is controversial. Aspirin and dipyridamole often are combined with plasma exchange but have not been shown conclusively to modify the course of TTP.8,64 Low-dose aspirin (e.g., 80 mg/day) has been suggested for thromboprophylaxis, once the platelet count exceeds 50 × 109/L.55
Transfusion of platelets may correlate with acute deterioration and death in TTP.7,29,65,66 Therefore, platelet transfusions are relatively contraindicated and should be reserved for the treatment of life-threatening hemorrhage, preferably after plasma exchange treatment has been initiated. Platelets generally need not be given prophylactically before establishing venous access for plasma exchange.67,68 Platelets have been transfused before emergency surgery, immediately after preparation by intensive plasma exchange.65
TTP that is refractory to plasma exchange usually responds to rituximab (e.g., 375 mg/m2 weekly for 4 doses). At least 80 percent of patients have complete responses within 1 to 3 weeks of starting treatment,69 including a normal ADAMTS13 level and disappearance of anti-ADAMTS13 antibodies (if present). Relapses occur in a minority of patients after successful treatment, usually after intervals of 6 months to 4 years, and most such patients respond to retreatment.
Acute reactions to rituximab are controlled by premedication with glucocorticoids, antihistamines, and analgesics. Because rituximab is removed by plasma exchange, it should be administered immediately after plasma exchange to maximize the interval until the next plasma exchange.
Rituximab has been given together with plasma exchange at the time of initial diagnosis, which may shorten the time to treatment response and reduce the incidence of relapse.55,69,70 Rituximab also has been administered preemptively to patients with persistent or recurrent severe ADAMTS13 deficiency after achieving remission of TTP, and this approach may prevent subsequent relapses.70,71
In some settings, rare but serious complications associated with rituximab have included bronchospasm, hypotension, serum sickness, susceptibility to infections, and progressive multifocal leukoencephalopathy.72 Such events have been very rare for patients with autoimmune diseases like TTP.73,74
Patients who have not been vaccinated for hepatitis B should be screened for hepatitis B infection before receiving rituximab. Those with evidence of past infection should be considered for antiviral prophylaxis as well as monitoring for hepatic injury and viral reactivation for 6 to 12 months after treatment.74
Splenectomy can result in lasting remissions or reduce the frequency of relapses for some patients with TTP that is refractory to plasma exchange or immunosuppressive therapy, presumably by removing a major site of anti-ADAMTS13 antibody production.75,76 Laparoscopic splenectomy can be performed safely in most patients regardless of platelet count.77
Anecdotal experience suggests that vincristine may be beneficial for refractory TTP, although its efficacy is difficult to assess. Dosing schedules have included 2 mg intravenously on day 1 followed by 1 mg on days 4 and 7,78 or 2 mg intravenously per week for 2 to 14 weeks.79 Prostacyclin analogues80,81 or high-dose intravenous immunoglobulins82,83 have been used without convincing evidence of efficacy.
Although cyclosporine can cause secondary thrombotic microangiopathy, apparent responses, with normalization of ADAMTS13 activity, have been observed with cyclosporine 2 to 3 mg/kg daily in two divided doses as an adjunct to plasma exchange.84
Other treatments have included oral or intravenous cyclophosphamide, oral azathioprine,55 bortezomib,85 mycophenolate,86 N-acetylcysteine,87 combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone,88 and autologous stem cell transplantation.89 Agents that prevent the binding of VWF to platelets are under development and may prove useful for the treatment of TTP.90,91
Daily laboratory monitoring should include complete blood count with platelet count, LDH, electrolytes, blood urea nitrogen and creatinine. Because of the high incidence of cardiac damage,29 continuous electrocardiographic monitoring and periodic assessment of cardiac enzymes should be considered. Patients should receive supplemental folic acid and vaccination for hepatitis B.55 Allergic reactions, metabolic alkalosis, and hypocalcemia associated with plasma exchange should be prevented or treated by appropriate adjustments in therapy.
After the platelet count increases to above 50 × 109/L, prophylaxis for venous thromboembolism may be instituted with low-molecular-weight heparin61 and low-dose aspirin.55
The platelet count normalizes after a median of 11 plasma exchanges, with a wide range of four to 55 sessions.92 Normalization of serum LDH lags behind the platelet count by approximately 9 days and persistent elevation of LDH does not correlate with the risk of exacerbation or relapse.93
Exacerbations are defined as TTP recurring within 30 days after a treatment response and 25 to 50 percent of patients have an acute exacerbation within 2 weeks that requires further treatment with plasma exchange. Some have repeated exacerbations over several months.94 A durable treatment response, lasting more than 30 days, is achieved eventually in approximately 80 percent of patients.92
Relapses, defined as recurrences more than 30 days after a complete response, occur in up to one-third of patients within 2 years after treatment with plasma exchange and glucocorticoids alone. Most relapses occur during the first year, but have occurred 13 years or more after diagnosis.26,94 Evaluation for relapsing TTP should be considered for any symptom compatible with thrombotic microangiopathy, especially in association with a common trigger of relapse such as infection, surgery, or pregnancy.34,95 Relapsing patients typically respond to plasma exchange. Relapses in TTP are associated with severe ADAMTS13 deficiency and detectable ADAMTS13 autoantibody inhibitors. Conversely, patients without severe ADAMTS13 deficiency at diagnosis rarely relapse (approximately 9 percent across several studies) (reviewed in Ref. 96).
Serious catheter-related complications of plasma exchange therapy occur in approximately 26 percent of patients with TTP and include pneumothorax and hemorrhage, cardiac perforation, venous thrombosis, catheter thrombosis, and bacterial or fungal infections.97
Hives or pruritic reactions to fresh-frozen plasma occur in one- to two-thirds of patients but usually can be managed by premedication with antihistamines. High-volume plasma exchange causes metabolic alkalosis and hypocalcemia, and may cause unintentional platelet removal. Serious complications attributable to plasma are less common, occurring in approximately 4 percent of patients, and include bronchospasm, anaphylaxis, hypotension, hypoxia, and serum sickness.97
The mortality rate for TTP treated with plasma exchange ranges from 10 to 20 percent. Most deaths occur within a few days after presentation, and almost all occur within the first month.7,8,26,94
Late sequelae of TTP may include long-term deficits in quality of life and cognition in many patients,98,99 severe persistent neurologic deficits in 5 to 13 percent,100 chronic renal insufficiency in up to 25 percent,100 and dialysis dependent renal failure in 3 to 8 percent of patients.100,101
CONGENITAL THROMBOTIC THROMBOCYTOPENIC PURPURA
Congenital TTP, or Upshaw-Schulman syndrome, refers to TTP that is caused by inherited deficiency of ADAMTS13.
Schulman and colleagues102 and Upshaw103 first described a congenital disorder resembling TTP characterized by autosomal recessive inheritance and chronic relapsing thrombotic microangiopathy from infancy. Congenital TTP, or Upshaw-Schulman syndrome, shared many features with acquired TTP in adults, including the consistent response to plasma.103
Congenital TTP is caused by homozygosity or compound heterozygosity for inactivating mutations in the ADAMTS13 gene19 on chromosome 9q34 (reviewed in Ref. 104). The mutations usually impair the synthesis or secretion of ADAMTS13. As yet no evidence convincingly indicates locus heterogeneity in congenital TTP.
Congenital TTP is autosomal recessive and affects the genders almost equally.105 The prevalence of congenital TTP is approximately one per million population in Japan106 and appears to be similar elsewhere. Congenital TTP accounts for a few percent of patients presenting with TTP.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


