- Diabetes accounts for 75–90% of excess coronary artery disease risk seen in people with diabetes and enhances the effects of other cardiovascular risk factors.
- A range of hemodynamic and metabolic factors contribute to macrovascular disease in diabetes.
- The link between glucose control and cardiovascular disease (CVD) is not as strong as that seen with microvascular complications of diabetes.
- There is clear in vitro and in vivo evidence that glucose exerts direct and indirect toxic effects on the vasculature.
- Glycemic memory describes the deferred long-term injurious effects of prior glycemic status; early glycemic control appears to be important to reduce vascular complications in subsequent decades.
- Specific insulin resistance pathways appear to contribute to atherogenesis in diabetes.
- The accumulation of advanced glycation end-products (AGEs) exerts pro-inflammatory and pro-fibrotic effects on the vasculature via receptor independent and receptor dependent effects. The AGE receptor (RAGE) is pivotally involved in the pathogenesis of diabetes accelerated atherosclerosis.
- The components of the classic renin angiotensin system (RAS) and in particular more recently discovered components such as angiotensin converting enzyme 2 appear to contribute to macrovascular disease in diabetes. Inhibitors of the RAS have consistently demonstrated reduced endothelial dysfunction and atherosclerosis in animal models via suppression of inflammation, fibrosis and oxidative stress. There is also strong clinical evidence for vasculoprotection by RAS blockade.
- Other vasoactive components such as endothelin and urotensin II are also likely to contribute to macrovascular complications in diabetes and interact with the RAS. Furthermore, the role of novel tumor necrosis factor (TNF) related ligands, such as TNF-related apoptosis inducing ligand and osteoprotegerin, and the complement system in atherosclerosis are currently under evaluation.
- Treatments that reduce oxidative stress and inhibit inflammation such as peroxisome proliferator activated receptor agonists have been shown to be anti-atherosclerotic in experimental studies with some evidence, albeit not uniform, also having been obtained in clinical studies.
- A multifactorial approach treating conventional cardiovascular risk factors as well as diabetes specific risk factors is currently viewed as the optimal strategy to reduce the burden of CVD in diabetes.
Epidemiology of diabetic macrovascular complications
Macrovascular complications develop in patients with type 1 (T1DM) [1–3] and type 2 diabetes mellitus (T2DM) [4–6]. This is of a particular concern as the increasing prevalence of diabetes now also affects adolescents and younger adults, thus promoting the earlier development of long-term cardiovascular complications. Even after adjusting for concomitant risk factors such as hypertension and hyperlipidemia there remains an excess risk for cardiovascular disease (CVD) in people with diabetes [7,8]. Indeed, diabetes itself accounts for 75–90% of the excess coronary artery disease (CAD) risk and enhances the effects of other cardiovascular risk factors. Death from stroke and myocardial infarction (MI) are the leading causes of mortality in T1DM and T2DM [2,9].
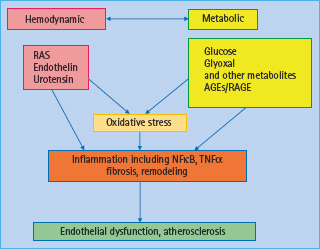
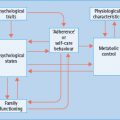
A range of hemodynamic and metabolic factors have been considered responsible for the development and progression of macrovascular disease in diabetes (Figure 39.1) [10]. In terms of hemodynamic factors, in particular the hormonal cascade known as the renin angiotensin system (RAS) has been shown to have a pivotal role in diabetes-associated atherosclerosis; however, other vasoactive hormone systems such as the endothelin (ET) [11] and urotensin systems [12,13] have also been implicated in diabetic macrovascular disease. More recently, a role for novel factors such as tumor necrosis factor (TNF) related apoptosis inducing ligand (TRAIL) [14] and the complement system has also been suggested [15].
Figure 39.1 Hemodynamic and metabolic mediators contribute to the pathogenesis of diabetic vascular complications. Hemodynamic factors include the renin angiotensin system (RAS), and other vasoactive factors. Metabolic factors include glucose and glucose metabolites such as the formation of advanced glycation end-products (AGEs). Both pathways interact with each other and lead to oxidative stress and inflammation, thus promoting endothelial dysfunction and atherosclerosis. NFκB, nuclear transcription factor κB TNF-α, tumor necrosis factor α.
Pathogenesis of diabetic macrovascular disease
Atherosclerosis is initiated by the adhesion of monocytes to the vascular wall, particularly to endothelial cells, followed by transmigration of monocytes into the subendothelial space [16]. Monocytes differentiate into macrophages which, via uptake of lipids, transform into foam cells and accumulate in the vascular wall. The early atherosclerotic process results in the formation of fatty streak lesions which over time develop into more advanced lesions, characterized by infiltration with vascular smooth muscle cells (VSMC), formation of a necrotic core and further lipid accumulation [16]. In humans, these lesions can demonstrate features of instability and plaque rupture including intraplaque hemorrhages as well as heightened thrombogenicity.
Role of hyperglycemia
A pivotal role for glycemic control and duration of diabetes exposure has been well established for microvascular complications in the UK Prospective Diabetes Study (UKPDS) study [4,17], although the data are not as convincing for macrovascular disease. The importance of glycemic control for macrovascular injury has only recently been extensively investigated in a series of studies that were reported over the last 12 months. It has been suggested that HbAlc acts as an independent and continuous risk factor for macrovascular disease [18–21]; however, this link is not as strong as has been identified with respect to microvascular complications. For an increase in HbAlc from 5.5% (37mmol/mol) to 9.5% (80 mmol/mol) there is a 10-fold increase in microvascular disease endpoints, whereas the risk for macrovascular disease endpoints increases only twofold.
Clinical trials and hyperglycemia
Recently, clinical trials such as the Action to Control Cardiovascular Risk in Diabetes trial (ACCORD) [22], Action in Diabetes and Vascular disease: PreterAx and DiamicroN-MR Controlled Evaluation (ADVANCE) trial [23] and the Veterans Affairs Diabetes Trial (VADT) [24], which explored tight glycemic control in patients with T2DM with established CVD, have investigated the association between tight glycemic control and cardiovascular endpoints. As observed in the initial reports of the Diabetes Control and Complications Trial (DCCT) in patients with T1DM and in the UKPDS trial in patients with T2DM, tight blood glucose control had little effect on macrovascular outcomes. This was further emphasized by the recent studies including the ADVANCE study [23] and ACCORD trial [22]. Both studies failed to demonstrate significant cardiovascular benefits, with the ACCORD study suggesting possibly deleterious cardiovascular outcomes in association with tight glucose control.
One possible explanation for the lack of a positive effect of tight glycemic control on cardiovascular outcomes may be the short duration of these trials (less than 5 years). Indeed, as best seen with respect to the longer follow-up of the Steno-2 study [25] and a recent long-term follow-up of the UKPDS [26], the benefits of prior intensified cardiovascular risk management including aggressive glucose control may not appear for up to 10 years after initiation of the studies. Thus, in view of the relative lack of an impact on CVD with intensive glucose control, other risk factor modification strategies need to be emphasized.
Direct and indirect glycotoxicity
Hyperglycemia is thought to have direct and indirect toxic effect on vascular cells. It has been suggested that increased glucose levels enter the polyol pathway at an increased flux rate leading to heightened formation of diacylglycerol. In addition, increased flux of glucose into the hexosamine pathway may contribute to glucose mediated vascular injury in diabetes. To investigate specifically hyperglycemia mediated atherosclerosis, aldose reductase, transgenic mice have been investigated [27]. These mice demonstrated increased glucose delivery via the polyol pathway and also showed early atherosclerosis, but did not develop advanced vascular lesions.
In vitro studies
Studies in cell culture experiments have obtained clear evidence that hyperglycemia induces a range of pro-atherogenic effects. Glucose directly activates monocytes–macrophages in vitro initiating increased expression of cytokines such as interleukin 1β (IL-1β) and IL-6 [28]. Furthermore, this leads to protein kinase C (PKC) and nuclear factor κB (NFκB) activation resulting in increased production of reactive oxygen species (ROS). Auto-oxidation of glucose can also lead to the formation of ROS and can mediate low density lipoprotein (LDL) oxidation. Scavenger receptors on activated macrophages can mediate the uptake of modified lipids such as the pro-atherogenic oxidized LDL. The formation of advanced glycation end-products (AGEs) in the hyperglycemic milieu can lead to the formation of modified albumin which inhibits scavenger receptor class B type 1 mediated efflux of cholesterol to high density lipoprotein (HDL). Therefore, prolonged hyperglycemia can indirectly lead to a range of secondary changes including changes in lipid profile, cellular lipid accumulation and foam cell formation as well as AGE mediated modification of proteins leading to altered cellular structure and function.
Animal models
The study of atherosclerosis in diabetes has long been hampered by the lack of an appropriate animal model. Our group and others have developed a murine model of diabetes associated macrovascular disease, the apoE knockout (KO) mouse rendered diabetic by multiple low doses of streptozotocin injections. This model is currently considered by the National Institutes of Health/Juvenile Diabetes Research Foundation (NIH/JDRF) co-sponsored Animal Models for Diabetes Complications Consortium (AMDCC) to be an appropriate model to study macrovascular disease in diabetes [29].
To address the separate roles of hyperglycemia and dyslipidemia in diabetes-associated atherosclerosis, mice deficient in the LDL receptor were bred with transgenic mice expressing a viral protein under control of the insulin promoter [30]. When infected with the virus, T cells mediate destruction of pancreatic β-cells that express the viral protein, thus closely mimicking the autoimmune response in human T1DM. In these animals, atherosclerosis development was accelerated even on a normal diet suggesting that hyperglycemia was driving atherosclerosis in these mice [30,31]. When animals were placed on a high fat diet, a further acceleration of atherosclerosis occurred suggesting that glucose and lipids may act through synergistic mechanisms to accelerate atherosclerosis. Furthermore, plaque disruption and intraplaque hemorrhages, features of plaque instability and potential plaque rupture were observed in this model [30].
Metabolic memory
The concept of “glycemic memory” describes the deferred effects of prior glycemic status on the subsequent development of diabetic complications. Episodes of poor glycemic control during the earlier stages of diabetes can precipitate or accelerate the development of complications later in the course of diabetes even when glycemic control is subsequently improved. Two studies have provided evidence for such a metabolic imprint, the DCCT/EDIC trial in T1DM [2] and the UKPDS in T2DM [26]. Both studies have shown that poor glycemic control was associated with an increased subsequent burden of complications. Another study in patients with T2DM, the VADT study, compared intensive with standard glucose control (6.9 vs. 8.4%, 52 vs 68 mmol/mol), but did not demonstrate significant cardiovascular protection after patients had been exposed to hyperglycemia for long periods of time (12–15 years) [24]. Therefore, it appears that early glycemic control is crucial for a long-term beneficial outcome on diabetic complications.
The concept of “metabolic memory” raises two important issues. First, hyperglycemia may expose patients to the harmful effects of hyperglycemia years before T2DM is diagnosed. Indeed, 25% of patients show complications at the time of diagnosis of T2DM. Therefore, it has been postulated that early diagnosis and strict glycemic control may be pivotal to reduce the induction of this metabolic memory with subsequent development of long-term diabetic vascular complications. Secondly, glycemic oscillations including peaks and troughs are not reflected in the HbA1c levels [32,33] but may have a key role in mediating growth factor and cytokine expression as well as inducing chromatin remodeling [34].
There are many cellular and molecular processes that may contribute to the mechanisms underlying metabolic memory with most of those relating to glycotoxicity. These pathways include the formation of AGEs, glycation of DNA, increased flux of glucose metabolism, leading to increased oxidative damage, overproduction of PKC-β as well as mitochondrial stress. Recently, it has been shown that transient hyperglycemia induces long-lasting activation of epigenetic changes in the promoter region of the NFκB subunit p65 in aortic endothelial cells both in vitro and in vivo. These hyperglycemia induced epigenetic changes and increased p65 expression were prevented by reducing mitochondrial superoxide production or superoxide induced generation of α-oxoaldehydes such as methylglyoxal (Figure 39.2) [34]. These studies suggest that transient hyperglycemia causes persistent atherogenic effects during subsequent normoglycemia by inducing long-lasting changes in chromatin remodeling. Specifically, hyperglycemia results in recruitment of the histone methyl transferase Set7 with increased H3K4 mono-methylation of the proximal promoter region of the NFκB subunit p65 gene leading to increased expression of pro-atherogenic pathways including monocyte chemotactic protein 1 (MCP-1) and vascular cell adhesion molecule 1 (VCAM-1) [34]. Subsequent studies in mice, which were initially diabetic but returned spontaneously to normoglycemia, have also demonstrated persistent upregulation of pro-inflammatory genes such as p65 and MCP-1 as a result of prior hyperglycemia. Furthermore, more detailed epigenetic studies revealed that changes in histone modifications as a result of prior hyperglycemia also included changes in H3K9 methylation of the p65 promoter as well as effects on various histone methyl transferases and interestingly also demethylases such as LSD-1 [35].
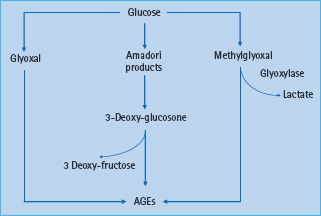
Figure 39.2 The formation of advanced glycation end-products (AGEs) can involve early glucose metabolites such as glyoxal and methylglyoxal, highly reactive dicarbonyls and key precursors of AGEs.
Insulin resistance
Insulin resistance occurs in T2DM and in patients with impaired glucose tolerance, and has been associated with increased cardiovascular risk [36–38]. There is now increasing evidence that insulin resistance promotes atherogenesis as an independent risk factor [39–41]. Furthermore, insulin resistance is often associated with a pro-atherogenic lipid profile that includes a high very low-density lipoprotein (VLDL) component, a low HDL and small dense LDL.
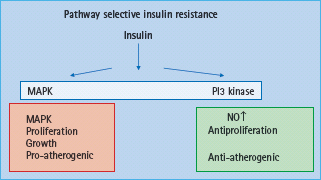
In vitro studies have shown that insulin exerts both pro-atherogenic and anti-atherogenic effects [42,43]. This has led to the hypothesis of pathway specific insulin resistance (Figure 39.3). It has been suggested that insulin resistance towards glucose transport also affects resistance to the antiproliferative effects of insulin, whereas the signaling pathways leading to cellular proliferation remain intact (Figure 39.3). Insulin signaling via phosphatidylinositol 3 kinase (PI3K) has been associated with antiproliferative and antithrombogenic effects with decreased adhesion molecule expression such as MCP-1, VCAM-1 and intercellular adhesion molecule 1 (ICAM-1). Furthermore, nitric oxide (NO) production is mediated via the PI3K signal transduction pathway and therefore is impaired in insulin resistance. NO has been shown to reduce LDL oxidation and proliferation of VSMC. In contrast, effects on VSMC proliferation and migration are mediated via the Ras/Raf/MEKK/MAPK signal transduction pathway which is further stimulated in the context of hyperinsulinemia, thus suggesting that pathway specific insulin resistance contributes to the pro-atherogenic effects of insulin. More recently, it has been shown that IL-6 decreases insulin stimulated NO production from endothelial cells via decreased activity of insulin signaling mediated by enhanced TNF-α production [44]. Paradoxically, IL-6 increases insulin stimulated glucose uptake into skeletal muscle and adipose tissue via enhanced insulin signaling, yet IL-6 and insulin have not been linked to increased TNF-α expression in skeletal muscle [44].
Figure 39.3 Pathway selective insulin resistance leading to endothelial dysfunction and atherosclerosis. MAPK, mitogen activated protein kinase; NO, nitric oxide; PI3 kinase, phosphatidylinositol 3 kinase.
Formation of AGE
AGE formation originates from early glycation products, the Schiff bases, which form the more stable Amadori products such as 1-amino-1-deoxyfructose derivatives. The Amadori products undergo further enzymatic modifications resulting in a number of reactive intermediates such as 3-deoxyglucosone and methyl-glyoxal (Figure 39.2). Methylglyoxal reacts with amino, sulfudryl and guanidine functional groups in proteins causing the browning, denaturation and redox active di-amine cross-linking between lysine residues of the target amino acids. Methylglyoxal also generates hydroimidazolones, N-E-(carboxyethyl) lysine, a homolog of carboxymethyl lysine (CML) and methylglyoxal lysine dimer [45,46]. The AGE-based cross-links are resistant to enzymatic degradation and therefore very stable [47]. The rate of AGE formation is dependent on multiple factors including the ambient concentrations of various sugars including glucose, the extent of oxidative stress and the duration of exposure to these various stimuli [48,49].
Direct effects of vascular AGE accumulation
There is evidence from experimental studies that AGEs directly influence endothelial function [50] as well as enhancing the evolution of macrovascular disease [51,52]. AGEs mediate their effects both directly and via receptor-mediated mechanisms. AGE accumulation in the vascular wall is associated with changes in the structural integrity of proteins, disturbance of their cellular function and degradation of these proteins [28]. AGEs accumulate on many proteins including collagen, albumin and apolipo-proteins. Furthermore, the cross-linking of AGEs with matrix molecules can disrupt matrix–matrix and matrix–cell interactions. It has been shown that AGE cross-linking to collagens decreases vascular elasticity and reduces vascular compliance resulting in increased vascular stiffness [53,54]. In addition, AGEs are also able to quench NO [50,55] and generate ROS by stimulating nicotinic acid adenine dinucleotide phosphate (NADPH) oxidase activity [56].
AGE binding proteins
The receptor-mediated effects of AGEs occur via binding to proteins such as RAGE [57], AGE-R1 (p60), AGE-R2 (p90) and AGE-R3 (galectin-3), the ezrin-radixin-moesin (ERM) family of proteins [58] macrophage scavenger receptor ScR-11 and CD-36 [59]. The exact roles of AGE-R1, AGE-R2 and AGE-R3 have not been fully elucidated. Finally, the interaction between AGEs and several macrophage scavenger receptors, such as CD36 [59], has been postulated to also promote atherosclerosis with studies using CD36-knockout mice supporting the view that CD36 promotes atherosclerosis [60].
Receptor for AGEs, RAGE
RAGE is a multiligand signal transduction receptor of the immunoglobulin superfamily of cell surface molecules that acts as a pattern recognition receptor [57]. In addition to binding ligands actively participating in inflammation and immune responses, RAGE serves as an endothelial adhesion receptor for leukocyte integrins and promotes leukocyte recruitment and extravasation of infiltrating cells. Of direct relevance to diabetic macrovascular complications, RAGE is found on endothelial cells and monocytes–macrophages [52,61,62], with RAGE having been implicated in inflammatory lesions in many disorders [63].
Downstream effects of RAGE activation
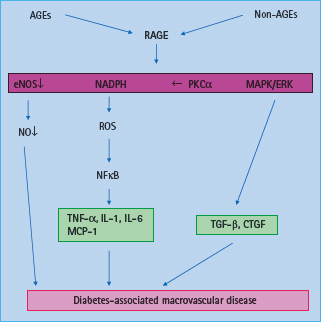
Engagement of RAGE leads to activation of the pro-inflammatory transcription factor NFkB (Figure 39.4) [64]. AGE binding to RAGE activates various signaling pathways, including NADPH oxidase [56], MAPKs, p21ras [65], extracellular signal-regulated kinases (ERKs) [66] and PKC causing activation and translocation of NFκB [64]. Furthermore, expression of RAGE itself can be induced by NFκB [67]. RAGE expression within tissues is markedly enhanced in response to metabolic disturbances such as diabetes, dyslipidemia, uremia and aging, possibly because of accumulation of AGEs in these conditions [61,63]. In plaques from diabetic apoE KO mice, upregulation of connective tissue growth factor (CTGF) has been demonstrated that appears to be AGE dependent [51] and may be mediated via RAGE. Intracellular accumulation of AGEs may also promote phenotypic conversion of VSMCs into foam cells within atherosclerotic plaques [68].
Figure 39.4 Activation of AGE receptor (RAGE) contributes to diabetes associated macrovascular disease via increased production of reactive oxygen species (ROS), decrease in nitric oxide (NO) availability, activation of the nuclear transcription factor κB (NFκB) and tumor necrosis factor α (TNF-α) as well as activation of profibrotic growth factors such as transforming growth factor β (TGF-β) and connective tissue growth factor (CTGF) associated with vascular remodeling. eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; MAPK, mitogen activated protein kinase; MCP, monocyte chemotactic protein; NADPH, nicotinic acid adenine dinucleotide phosphate; PKC, protein kinase C.
Studies reducing vascular AGE accumulation
A variety of pharmacologic interventions have been used to reduce the accumulation of AGEs via decreasing the total AGE load or via chemical modification of existing AGEs into inactive forms [69,70]. Aminoguanidine is a potent inhibitor of the formation of AGEs, scavenges reactive dicarbonyl AGE precursors [69,70] and decreases oxidative damage to mitochondrial proteins [71]. In experimental and clinical diabetes, aminoguanidine treatment has been shown to reduce microvascular [72] and more recently macrovascular complications in a model of accelerated atherosclerosis, the diabetic apoE KO mouse [51].
Another potential agent which inhibits AGE accumulation is ALT-711 or alagebrium [73]. Based initially on a range of in vitro studies, this thiazolium compound and its original prototype, phenylthiazolium bromide [74], have been shown to cleave pre-formed AGEs and thus one of the postulated mechanisms of ALT-711 is as an AGE cross-link breaker [73].
The removal of established cross-links in diabetic rats with ALT-711 has been shown to be associated with reversal of the diabetes-induced increase in large artery stiffness, increased collagen solubility and reduced vascular and cardiac AGE accumulation [54,75–77]. In addition, alagebrium prevents the progression of nephropathy [78,79] possibly via direct inhibition of PKC-α phosphorylation [80], thus reducing renal expression of vascular endothelial growth factor (VEGF). Treatment with ALT 711 in diabetic apoE KO mice was associated with a significant reduction in atherosclerosis [51] This anti-atherosclerotic effect was associated with reduced vascular AGE accumulation and RAGE expression. Alagebrium treatment was also associated with less inflammation and reduced expression of pro-fibrotic growth factors, in particular CTGF [81,82] In the clinical setting, ALT 711 has been shown to reduce pulse pressure and improve vascular compliance in patients with systolic hypertension [54].
Soluble RAGE
There are three major splice variants of RAGE [83]. First, there is the full-length RAGE receptor; secondly, the N-terminal variant, that does not contain the AGE-binding domain; and, thirdly, a C-terminal splice variant, soluble RAGE (sRAGE), which does not contain the trans-membrane and effector domains. It remains controversial as to whether the effects of sRAGE are primarily as a decoy to ligands such as AGEs or whether sRAGE acts as a competitive antagonist to the full-length biologically active RAGE [83,84].
Soluble RAGE and diabetes associated atherosclerosis
Soluble RAGE (sRAGE) has been identified as having therapeutic value in a model of diabetes associated atherosclerosis, the streptozotocin diabetic Apo E KO mouse [85,86]. In the original study, Park et al. [85] reported that diabetic apoE KO mice treated with sRAGE showed a dose-dependent suppression of atherosclerosis and reduced plaque complexity. These beneficial effects were independent of effects on glucose or lipid levels. Furthermore, AGE levels in these diabetic mice were suppressed in a dose-dependent manner by sRAGE to levels similar to those seen in non-diabetic animals. Furthermore, the effect of RAGE blockade with sRAGE was investigated in established atherosclerosis [86]. Administration of sRAGE decreased the expression of RAGE, as well as reducing the number of infiltrating inflammatory cells and gene expression for transforming growth factor β (TGF-β), fibronectin and type IV collagen in both the aorta and the kidney in association with reduced plaque area. Therefore, it was concluded by these investigators that RAGE activation not only contributes to lesion formation, but also to the progression of atherosclerosis.
Soluble RAGE treatment has also been reported to be effective in reducing vascular complications in other models of atherosclerosis and diabetes. Atherosclerosis in the LDL receptor –/– mouse made diabetic by streptozotocin injection [87] was significantly attenuated by sRAGE treatment. ApoE KO mice bred onto a db/db background, a model of T2DM and deficient leptin receptor signaling, showed increased atherosclerosis which was significantly attenuated by daily sRAGE treatment [88].
To understand better the specific role of RAGE in the genesis of vascular lesions, animals selectively deficient in RAGE/apoE (RAGE –/–) have been created [89,90] These mice completely lack not only tissue bound full-length RAGE, but also soluble RAGE. As had been predicted by the pharmacologic intervention studies directed towards the RAGE ligands and AGEs, RAGE-knockout mice bred onto an apoE–/– background showed a marked reduction in plaque area in the presence and absence of diabetes [89,90]. More recently, the effect of RAGE deletion on atherosclerosis development has been investigated in streptozotocin diabetic RAGE/apoE KO mice and showed a significant reduction in atherosclerotic plaque area when compared with diabetic apoE KO mice expressing RAGE. These vascular changes seen in the streptozotocin diabetic double RAGE/apoE KO mice were associated with reduced inflammation, less accumulation of RAGE ligands such as S100/CML, decreased infiltration by macrophages and T lymphocytes and a reduction in expression of pro-fibrotic and pro-inflammatory growth factors and cytokines [90].
These promising results with RAGE antagonism have encouraged the development of RAGE neutralizing compounds for clinical use. The RAGE modulating agent TTP488 is currently being considered in phase II clinical trials in patients with Alzheimer disease and in patients with diabetic nephropathy. Another compound, TP4000, will soon enter phase I clinical trials (www.Clinicaltrials.gov and www.ttpharma.com); however, currently, the major focus of antagonizing RAGE does not appear to involve studies addressing CVD in the absence or presence of diabetes.
Interaction with the renin angiotensin system
Because diabetic complications appear to be multifactorial in origin and involve interactions between hemodynamic pathways such as the RAS and metabolic pathways such as hyperglycemia and the formation of AGEs, there has been increasing investigation of the potential links between these various pathways (Figure 39.1) [91]. There is increasing evidence that AGE accumulation can induce an upregulation of certain components of the RAS, although these studies have been performed predominantly in the renal context [92]. Furthermore, angiotensin-converting enzyme (ACE) inhibition has been reported to confer its end-organ protective effect, partly via a reduction in AGEs and an increase in sRAGE [93]. Therefore, the status of the RAS could represent a key modulator in AGE-induced diseases. More recently, other therapeutic interventions shown to be anti-atherosclerotic have been reported to exert part of their vasculoprotective effect via inhibition of the AGE/RAGE pathway. These include angiotensin II receptor blockers (ARBs), peroxisome proliferator-activated receptor α (PPAR-α) and PPAR-γ agonists and statins [94–96].
Role of vasoactive hormones in diabetes-related atherosclerosis
Classic RAS
Renin was described more than 100 years ago by Tigerstedt & Bergman [97]. Nevertheless, our understanding of the RAS is still not complete and has grown increasingly complex over the last decade. The classic pathway is now very well characterized (Figure 39.5). The generation of angiotensin (AT) I and II is not restricted to the systemic circulation but this production also takes place in vascular and other tissues (Figure 39.5) [98].
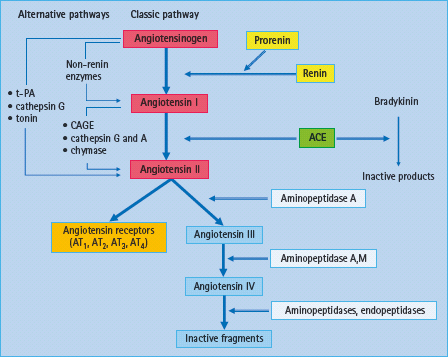
Figure 39.5 Overview of the enzymatic cascade of the renin angiotensin system (RAS): classic and alternative pathways. In the classic pathway, renin cleaves the decapeptide angiotensin I from angiotensinogen. Angiotensin I is then converted to angiotensin II which acts through several receptor subtypes, being AT1 and AT2 receptors the more relevant in the vasculature. CAGE, chymostatin-sensitive angiotensin II-generated enzyme; t-PA, tissue plasminogen activator
Novel aspects of the RAS: ACE2
In 2000, a further enzyme associated with the generation of AT peptides was identified – ACE2, a carboxypeptidase with sequence similarity to ACE [99]. ACE2 does not generate AT II but increases the formation of AT 1-7 (Figure 39.6). This heptapeptide causes vasodilatation and has growth inhibitory effects [100]. Further research, including into specific inhibitors, is needed to understand the many functions of ACE2 inside and outside the RAS.
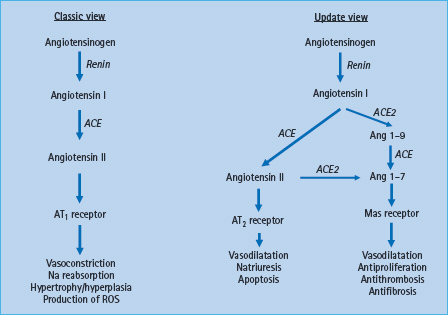
Figure 39.6 New aspects of the renin angiotensin system (RAS) components and its interactions. The classic RAS illustrates the main pathway for angiotensin II (AT II) generation from angiotensin I (AT I) via ACE, with effects being mediated via AT1 receptor. The updated view illustrates the new components of the RAS in which ACE2 has a role to degrade AT I to AT 1-9, and AT II to the vasodilatator AT 1-7 which acts through the Mas receptor.
AT receptors
The effects of all AT peptides are mediated through specific cell surface receptors (Figures 39.5 and 39.6). The AT1 receptor mediates most of the effects usually associated with AT II. The role of the AT2 receptor subtype remains controversial, but it appears to antagonise certain effects of AT II mediated via the AT1 receptor [101].
Role of the RAS in macrovascular disease
It is now recognized that local RAS activation has an important role in the pathogenesis of diabetes-induced endothelial dysfunction and atherosclerosis [102–104]. As a result, there is a strong rationale for blockade of the RAS to prevent cardiovascular events in patients with diabetes. In addition, several recent clinical trials [105–110] have suggested that blockade of the RAS may protect against the development of T2DM in at-risk patients.
RAS activation and endothelial dysfunction
Endothelial dysfunction has been considered as an imbalance between vasoconstrictors and vasodilatators in the vascular tone [111] associated with a decrease in NO activity and increased AT II and endothelin (ET) activity [112]. Endothelial dysfunction is characterized by defective endothelium-dependent vasorelaxation in association with inflammatory cytokine-induced increased interactions with blood leukocytes, in which adhesion molecules such as VCAM-1 and chemo-attractants such as MCP-1 are essential [113]. Endothelial dysfunction is strongly associated with atherosclerosis (Figure 39.7) [114,115].
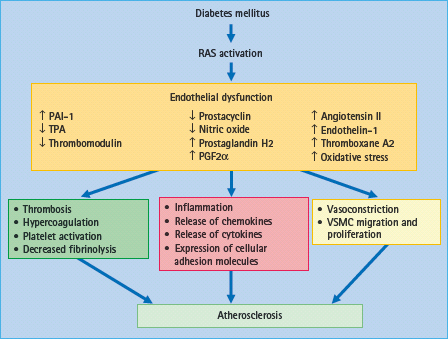
Figure 39.7 Effects of diabetes-induced renin angiotensin system (RAS) activation on mechanisms associated with endothelial dysfunction and atherosclerosis. In diabetes, activation of RAS induces endothelial dysfunction which is characterized by vasoconstriction, inflammation, cellular growth and thrombosis. By losing its protective properties, dysfunctional endothelium is a major promoter of atherogenesis and, consequently, cardiovascular events. PAI-1, plasminogen activator inhibitor-1; TPA, tissue plasminogen activator; VSMC, vascular smooth muscle cells.
Crucial to the pathogenesis of diabetes-induced endothelial dysfunction in the early stages, in the absence of structural changes of the vessel wall, is the activation of the RAS. Although measurements of components of the RAS in plasma have, in general, suggested suppression of this system in diabetes, there is increasing evidence for activation of the local cardiovascular RAS in the diabetic context [116,117]. It has been demonstrated that there is an increase in ACE expression and activity, in AT II expression, and in AT1 receptor expression in the aortic wall of diabetic apoE KO mice suggesting a key role for RAS activation in the pathogenesis of diabetes associated endothelial dysfunction [102,118].
RAS and regenerative endothelial cell repair
A role for the RAS has also been demonstrated in affecting the number of regenerative endothelial progenitor cells in patients with diabetes. An increased concentration of circulating endothelial progenitor cells, which are believed to maintain the integrity of the vascular endothelium, has been associated with a favorable cardiovascular outcome in patients with CAD [119]. A study in patients with T2DM [120] suggested that treatment with an ARB (olmesartan) increases the number of regenerative endothelial progenitor cells, which could contribute to the beneficial cardiovascular effects seen with AT1 receptor blockade.
Role of AT 1-7 and ACE2 in endothelial dysfunction
It has been hypothesized that disruption of the ACE–ACE2 balance may result in abnormal blood pressure, with increased ACE2 expression protecting against hypertension, and ACE2 deficiency causing hypertension (Figure 39.6) [121]. As reported above, it is well established that AT II produces endothelial dysfunction through different pathways, such as increasing oxidative stress and exerting proliferative and pro-thrombotic activities [122]. Recently, it has been observed that AT 1-7, which is generated by the enzyme ACE2, promotes the release of NO and prostaglandins [123,124] and potentiates bradykinin effects in different experimental models [124,125]. In addition, AT 1-7 inhibits growth of VSMC [126], platelet aggregation and thrombosis [127], inflammation, fibrosis [128] and oxidative stress [129] which in turn might lead to restoration of endothelial function. It appears that AT 1-7 can antagonize AT II effects, not only by the stimulation of other vasodilatators but also through AT1 receptor inhibition. In this regard, Kostenis et al. [130] reported that the Mas receptor, which is increasingly considered to be the major receptor conferring the biologic effects of AT 1-7, seems to act as a physiologic antagonist of AT1 receptor and thus counteracting many of the actions of AT II at the endothelial level.
RAS activation and atherosclerosis
Clinical and experimental evidence clearly indicates that activation of the RAS is central to almost all these pro-atherosclerotic pathways (Figure 39.7) [103,131]. Diet et al. [104] observed increased ACE protein accumulation within the atherosclerotic plaque in human coronary arteries suggesting that ACE may contribute to an increased production of local AT II which may participate in the pathophysiology of artery disease. In addition, there is evidence that AT II and the AT1 receptor are overexpressed in atherosclerotic plaques [102,132,133]. Indeed, blockade of the RAS with an ACE inhibitor or an AT1 receptor antagonist prevents atherosclerosis by mechanisms involving inhibition of pro-inflammatory molecules such as VCAM-1 and MCP-1 and pro-sclerotic and pro-proliferative cytokines such as CTGF and platelet derived growth factor. These observations confirm a key role for RAS activation in the development and progression of atherosclerosis.
Production of pro-inflammatory cytokines, such as IL-1β, TNF-α and IL-6, have a major role in the pathogenesis of atherosclerosis [134]. IL-6, ACE and AT1 receptors have been detected in stable and unstable atherosclerotic plaques [132,135]. AT II stimulates the redox sensitive nuclear transcription factor, NFκB, which could serve as a unifying signaling system for inflammatory stimuli in atherogenesis through enhanced expression of adhesion molecules ICAM-1 and VCAM-1, E-selectin, MCP-1 and IL-8. Several clinical and experimental studies have demonstrated that both ACE inhibitors and AT1 receptor blockers decrease the expression of several adhesion molecules, thus confirming that the chronic inflammatory response associated with atherosclerosis appears to be modulated by AT II at every level and can be targeted therapeutically by RAS inhibition [103,116,136].
Moreover, the sustained pro-inflammatory state seems to play an important part in the transformation of a stable atherosclerotic plaque into a vulnerable plaque prone to rupture. Plaque rupture has been connected with activation of matrix metalloproteinases in the fibrous cap of the atherosclerotic lesion [137], and there is evidence that AT II is implicated in matrix metalloproteinase activation, both through a direct action and through induction of pro-inflammatory cytokines such as IL-6.
ACE2 and diabetes accelerated atherosclerosis
Although studies on the expression and activity of ACE2 in atherosclerosis are limited, there is increasing evidence that such more recently identified components of the RAS may be involved in the development and progression of atherosclerosis. Recently, Zulli et al. [138] have shown very high expression of ACE2 in endothelial cells, macrophages and α-smooth muscle cells within atherosclerotic plaques in a rabbit model of atherosclerosis. It is not clear whether this increase in ACE2 is in response to injury in an attempt to protect the vessel by increasing levels of AT 1-7.
In the human context, there is evidence for a significant activation of the cardiac RAS after coronary artery occlusion, and it is well known that RAS blockade reduces remodeling and improves survival in humans after an MI. In recent studies [139], cardiac ACE2 expression and activity are also increased with experimental MI. All these observations suggest that an imbalance in the RAS activity has a central role in the pathogenesis of atherosclerosis. Nevertheless, as yet, it is unknown if deficiency of ACE2 contributes to the accelerated atherosclerotic disease process seen in diabetes.
RAS and oxidative stress
There is increasing evidence that the local production of ROS has a pivotal role in atherosclerosis, specifically in the diabetic milieu. Increased vascular superoxide production in aortas from diabetic atherosclerotic apoE KO mice has been demonstrated [94]. These changes were mediated by increased NAD(P)H oxidase (Nox) activity in the aorta with increased expression of various Nox subunits including p47phox, gp91phox and rac-1 [94]. Evidence of increased local vascular ROS generation in diabetes is further supported by the demonstration of increased nitrotyrosine staining in these diabetic plaques. Indeed, interventions that reduce vascular superoxide production such as PPAR-α and PPAR-γ agonists have been associated with reduced plaque formation, further emphasizing the link between vascular oxidative stress and atherosclerosis [94,140]. Furthermore, the deletion of anti-oxidant enzymes such as glutathione peroxidase, specifically the Gpx1 isoform in the vascular wall, results in an increase in plaque area particularly in the diabetic context via increases in inflammatory mediators including adhesion molecules and chemokines [141]. More recently, it has been shown that treatment of GPx1 deficient mice with the Gpx1 analog, ebselen, reduced oxidative stress variables and atherosclerosis in diabetic GPx1/apoE KO mice [142]. Furthermore, in the Gpx1/apo E double KO mice, there was associated upregulation of RAGE further linking vascular RAGE expression to increased oxidative stress and accelerated atherosclerosis in settings such as diabetes [141].
Therapeutic implications
RAS blockade and endothelial dysfunction
As a result of previous observations, RAS blockade has emerged as an obvious and attractive therapeutic target. Early evidence for a clinical beneficial effect of inhibition of this system on impaired endothelial function was derived from the Trial on Reversing Endothelial Dysfunction (TREND) [143] which showed that ACE inhibition improves endothelial function in subjects with coronary artery disease. Moreover, O’Driscoll et al. [144] observed that ACE inhibition with enalapril improved both basal and stimulated NO-dependent endothelial function in normotensive patients with T2DM.
Data are also available for the role of ARBs in improving endothelial function such as the AT1 receptor antagonist, losartan [145,146]. Furthermore, in patients with T1DM treated with the ACE inhibitor ramipril or the ARB losartan for 3 weeks [147] improved endothelial dysfunction potentially mediated by increased bradykinin levels with ACE inhibition or increased levels of AT 1-7 levels.
RAS inhibition and cardiovascular protection
There is accumulative evidence that pharmacologic therapy that interrupts the RAS may afford special benefits in reducing CVD in people with diabetes [109,148,149]. In a post hoc subgroup analysis of the Captopril Prevention Project (CAPP) study, the patients with diabetes treated with captopril fared significantly better compared with those treated with conventional therapy (beta-blockers and diuretics) in terms of primary endpoint as well as for MI, all cardiac events and total mortality [148]. These relative beneficial effects of ACE inhibitor therapy were particularly striking in those at highest risk, specifically those with the highest median fasting glucose or those with more elevated blood pressure (BP). This is in contrast to the UKPDS [150], in which there was comparable CVD benefits for patients with T2DM who were randomized to captopril or atenolol, perhaps reflecting a lower CVD risk in the newly diagnosed patients with diabetes studied in the UKPDS.
In a sub-study of the Hypertensive Old People in Edinburgh (HOPE) study, the MICRO-HOPE [151], of the relative risk reduction in the 3577 patients who had diabetes and one other CVD risk factor, there was a risk reduction of 25% for combined CVD events, 37% for CVD mortality, 22% for MI and 33% for stroke. In the Fosinopril versus Amlodipine Cardiovascular Events Trial (FACET) study, the incidence of CVD events was less in hypertensive patients with T2DM treated with fosinopril than the amlodipine-treated group [152]. The European Trial on Reduction of Cardiac Events with Perindopril in Stable Coronary Artery Disease (EUROPA) [153] has shown that ACE inhibition reduced cardiovascular mortality and morbidity in patients with established CAD without left ventricular dysfunction. The data from these trials were pooled with those of the Quinapril Ischemic Event Trial (QUIET) [154] study in a meta-analysis that included a total of 31 555 patients [155]. This analysis showed that, compared with placebo, ACE inhibitor therapy produced a significant 14% reduction in all-cause mortality and MI, a 23% reduction in stroke and a 7% statistically significant reduction in revascularization procedures. Recently, the ADVANCE study [23] has shown that the administration of the ACE inhibitor perindopril and the diuretic indapamide in high-risk patients with T2DM induced a reduction in macrovascular outcome when compared with placebo.
Recent reports of trials with ARBs indicate that these agents have CVD protection effects in patients with T2DM similar to those observed with ACE inhibitors. In the Losartan Intervention For Endpoint reduction in hypertension (LIFE) study [109], treatment with losartan in patients with T2DM and left ventricular hypertrophy (LVH) resulted in a significant reduction in death from CVD and in particular all cause mortality. An analysis from the large Reduction of Endpoints in NIDDM with Angiotensin II Antagonist Losartan (RENAAL) study has shown that treatment with losartan in patients with T2DM, nephropathy and LVH reduced the cardiovascular risk to levels similar to those observed in patients without LVH [156]. Recently, the Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET) has shown that the ARB telmisartan provides a benefit similar to that of a proven ACE inhibitor such as ramipril in high risk patients with CVD or subjects with diabetes who have end-organ damage. This is a population similar to that examined previously in the HOPE study [157]. Furthermore, in the ONTARGET study the combination of ramipril with telmisartan, despite the further lowering of BP, did not reduce the risk of cardiovascular events, compared with an ACE inhibitor alone, but was associated with additional adverse effects including hypotension and renal dysfunction. Thus, an ACE inhibitor, or possibly an ARB, is an initial antihypertensive agent of choice in patients with diabetes because these agents have been shown to have the capacity to prevent or retard the development of diabetic macrovascular complications, thus significantly reducing cardiovascular mortality and morbidity.
The endothelin system
Endothelin (ET) was first discovered in 1988 by Yanagisawa et al. [158] and is one of the most potent vasoconstrictors known. There are three distinct ET genes that encode different mature ET sequences, designated ET-1, ET-2 and ET-3. Big ET-1 is converted into mature 21 amino acid ET-1 by ET-converting enzyme. ET-1 is predominantly present in endothelial cells. In 1990, the ETA and ETB receptor subtypes [159], were cloned. ETA receptors are found in VSMCs and mediate vasoconstriction and cell proliferation (Figure 39.8). ETB receptors are found in endothelial cells (ETB1) where they mediate vasodilation via the release of NO and on smooth muscle cells, where they may elicit vessel contraction and cell proliferation (Figure 39.8).
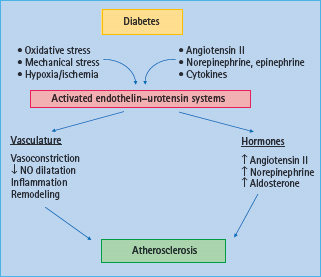
Figure 39.8 Proposed mechanisms of the endothelin and urotensin systems on atherosclerosis in diabetes. In diabetes, activation of the endothelin system induces vasoconstriction, vascular smooth muscle cells proliferation, wall thickening, inflammation and tissue remodeling thus leading to the development and progression of atherosclerosis.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


