- Type 2 diabetes (T2DM) is a multifactorial heterogeneous disease with a complex interplay of genetic and environmental factors influencing intermediate traits such as β-cell mass and development, insulin secretion and action, and fat distribution.
- Three general approaches to identifying genetic susceptibility factors are used: the study of candidate genes selected as having a plausible role in glucose homeostasis; a genome-wide scan to detect chromosomal regions with linkage in nuclear families; a genome-wide scan for association with common variants (single nucleotide polymorphisms and copy number variant); and the study of spontaneous, bred or transgenic animal models of T2DM.
- Common T2DM shows familial clustering, but does not segregate in a classic Mendelian fashion. It is thought to be polygenic and probably multigenic (many different gene combinations among people with diabetes). Only a small fraction of the genetic risk for common adult T2DM is known.
- Common nucleotide variants within or near the genes implicated in monogenic forms of diabetes (e.g. GCK, HNF4A or HNF1B/TCF2 in maturity-onset diabetes of the young [MODY], or WFS1 in Wolfram syndrome) may contribute to T2DM with modest risk effects.
- Candidate genes identified as having an association with common polygenic T2DM include the insulin promoter, class III alleles of the variable region upstream of the insulin gene, the peroxisome proliferator-activated receptor γ (PPARG), KCNJ11 encoding the pore forming Kir6.2 subunit of the p-cell inwardly rectifying KATP channel. Minor susceptibility might operate in some populations from other genes, including insulin receptor substrate 1 (IRS-1), adiponectin (ACDC) or ectonucleotide pyrophosphatase/phosphodiesterase 1 enzyme (ENPP1) in a context of obesity or diabesity.
- In genome scans of diabetic families, loci for T2DM have been found at several sites, including chromosomes 1q, 2q (NIDDM1), 2p, 3q, 12q, 11q, 10q and 20. NIDDM1 has been identified as coding for calpain 10, a non-Iysosomal cysteine protease with actions at the mitochondria and plasma membrane, and also in pancreatic β-cell apoptosis.
- In 2007, five large genome-wide association studies in European descent populations have identified new potential T2DM genes, including the Wnt signaling related transcription factors TCF7L2 and HHEX, the zinc transporter ZnT8 (SLC30A8), the CDK5 regulatory subunit-associated protein 1- I ike 1 (CDKAL1) and a regulatory protein for IGF2 (IGF2BP2). A consensus of close to 20 confirmed T2DM-susceptibility loci to date provided novel insights into the biology of T2DM and glucose homeostasis, but individually with a relatively small genetic effect. Importantly, these genes implicate several pathways involved in β-cell development and function.
- Compared with clinical risk factors alone, the inclusion of common genetic variants (at least those identified to date) associated with the risk of T2DM has a small effect on the ability to predict future development of T2DM. At the individual level, however, a combined genotype score based on 15 risk alleles confers a 5–8 fold increased risk of developing T2DM. Identifying the subgroups of individuals at higher risk is important to target these subjects with more effective preventative measures.
Genetic architecture of type 2 diabetes: a complex interaction of genetic susceptibility and environmental exposures
Type 2 diabetes mellitus (T2DM) is a heterogeneous metabolic disease resulting from defects of both insulin secretion and action [1,2]. The prevalence of diabetes has been estimated as 12.3% of the population of the USA, and worldwide to affect 285 million people with a projection to increase to 435 million by 2030 [3]; the vast majority of these individuals have T2DM. The disease affects various groups differently; minority racial groups including Hispanics, African-Americans, Native Americans or people living in the Middle East are affected at a higher rate than white individuals.
The etiology of T2DM is multifactorial, including genetic as well as prenatal and postnatal factors that influence several defects of glucose homeostasis, primarily in β-cell function and also in muscle and liver. It is generally accepted that T2DM results from a complex interplay of genetic and environmental factors influencing a number of intermediate traits of relevance to the diabetic phenotype (β-cell mass, insulin secretion, insulin action, fat distribution and obesity) [3]. Quantitative phenotypes related to glucose homeostasis are also known to be heritable, with a greater relative impact of genetic components on in vivo insulin secretion [4].
Although several monogenic forms of diabetes have been identified (see Chapter 15), such as maturity-onset diabetes of the young (MODY) and maternally inherited diabetes and deafness (MIDD) [5,6], diabetes in adulthood seems to be a polygenic disorder in the majority of cases. T2DM shows a clear familial aggregation, with a risk for people with familial diabetes that is increased by a factor of 2–6 compared with those without familial diabetes; it does not segregate in a classical Mendelian fashion, and appears to result from several combined gene defects, or from the simultaneous action of several susceptibility alleles, or else from combinations of frequent variants at several loci that may have deleterious effects when predisposing environmental factors are present [3,7], T2DM is probably also multigenic, meaning that many different combinations of gene defects may exist among subgroups of people with diabetes. Whereas the current worldwide epidemic of T2DM is greatly driven by lifestyle and dietary changes, a combination of these environmental factors and susceptible genetic determinants contribute to the development of T2DM [7].
The primary biochemical events leading to common diabetes in adulthood are still largely unknown in most cases, even if new genetic and biologic insights into T2DM etiology have recently arisen from the completion of genome-wide association (GWA) studies in several European descent populations [3,7]. Genetic and environmental factors may affect both insulin secretion and insulin action [1], and a variety of environmental factors can be implicated in the clinical expression of T2DM (see Chapter 4), such as the degree and type of obesity, sedentary lifestyle, malnutrition in fetal and perinatal periods, lifestyle and different kinds of drugs such as steroids, diuretics and antihypertensive agents (see Chapter 16). It is noteworthy that obesity, which is one of the so-called risk factors of T2DM, is also clearly under genetic control. Both disorders are frequently associated and share many metabolic abnormalities, which suggests that they might also share susceptibility genes [8,9]. Moreover, retrospective studies have shown that low birth weight is associated with insulin resistance and T2DM in adulthood [10,11]. It has been proposed that this association results from a metabolic adaptation to poor fetal nutrition [12]. The identification of gene variants that contribute both to variation in fetal growth and to the susceptibility to T2DM, however, suggests that this metabolic “programming” could also be partly genetically determined [13].
These complex interactions between genes and environment complicate the task of identifying any single genetic susceptibility factor for T2DM. Three general approaches have been adopted to search for genes underlying complex traits such as T2DM [6], as described in Table 12.1.
Table 12.1 Genetic and genomic approaches used for the study of type 2 diabetes genes.
| Family-based studies |
| Linkage studies (genome-wide scan with microsatellite markers) Transmission disequilibrium test (in sibships or nuclear families) Study of monogenic diabetes (extreme phenotypes, syndromic diseases) |
| Population-based studies |
| Survey of candidate genes and targeted genomic regions ( ~100 SNPs) Genome-wide association study (HapMap-based SNPs choice, 500K–1M SNPs) |
| Other types of genetic markers (rare variants, CNVs, insertion/deletion) Study of general populations and prospective cohorts (epidemiology-based data) |
| Functional genomic approaches |
| Gene expression profi ling, transcriptomics (microarray gene expression data in human and animal models) |
| Molecular and cellular analyses ( in vitro and in vivo model systems) |
| Expressed quantitative trait linkage (in human and animal models) |
| Epigenetics (in humans, and animal models) |
CNV, copy number variant; SNP, single nucleotide polymorphism.
The first approach was to focus on candidate genes, that is, genes selected as having a plausible role in the control of glucose homeostasis and/or insulin secretion, on the basis of their known or presumed biologic functions. Although this approach has led to the identification of several susceptibility genes with small effects (see below), no genes with a moderate or major effect on the polygenic forms of diabetes have been found. Possible explanations for this failure to identify genes with a major effect include the possibility that they do not exist, or our incomplete knowledge of the pathophysiologic mechanisms of T2DM and the genes that control them, has obscured the choice of candidates.
The second approach is to perform genome-wide scans for linkage in collections of nuclear families or sib-pairs with T2DM [14], or for GWA in large case–control samples, as was performed in recent years [15]. These “hypothesis-freen” approaches require no presumptions as to the function of the susceptibility loci. Although a large number of genomic regions with presumed linkage have been mapped [11–14], identification of the susceptibility genes and causal variants within these regions has proceeded at a very slow pace. In contrast, the recently completed GWA studies from several independent European case–control cohorts have released at least a dozen of confirmed at-risk loci, with unexpected susceptibility genes for T2DM and a number of at-risk variants having been replicated in most of the European populations investigated [6,15,16].
A third approach has used microarray gene expression analysis in attempt to define genetic alterations in T2DM. Through such analysis, a defect in skeletal muscle of people with diabetes was discovered and characterized by a coordinated decrease in the expression of nuclear-encoded genes involved in mitochondrial oxidative phosphorylation [17]. This defect appears to be secondary to reduced expression of the transcriptional coactivators PGC-1α and PGC-1β. Similar changes in expression have been observed in some cohorts of first-degree relatives of individuals with diabetes, suggesting that these may be heritable traits [18]. A second potential gene is the transcription factor ARNT/HIF1β, which was identified using islets isolated from individuals with T2DM compared with normal glucose-tolerant controls. Reduced ARNT levels in human diabetic islets were associated with altered β-cell expression of other genes involved in glucose sensing, insulin signaling and transcriptional control [19], suggesting an important role for ARNT in the impaired islet function of human T2DM.
Another complementary approach to identifying diabetes genes is to study spontaneous (such as the Zucker diabetic fatty rats or fat/fat mice, [20]), bred (like the GK rats, [21]) or transgenic (using β-cell-specific gene inactivation, [22–24]) animal models of T2DM. The genes responsible for diabetes in these models may not necessarily be major players in typical T2DM in humans, but such studies provide the most direct way of improving the overall understanding of the molecular circuitry that maintains glucose homeostasis. Nevertheless, despite the evidence of a strong genetic background in T2DM, very little is yet known about the genetic risk factors for T2DM. By far the most striking results in defining etiologic genes have been obtained by studying the highly familial MODY form of young-onset diabetes or other rare forms of monogenic diabetes.
Genes involved in monogenic diabetes and their relevance to adult-onset T2DM
Some of the most compelling evidence that inherited gene defects can cause glycemic dysregulation comes from the clinical and genetic description of monogenic forms of diabetes, including MODY characterized by an autosomal dominant inheritance of young-onset diabetes, neonatal or early infancy diabetes, or diabetes with extrapancreatic features (see Chapter 15) [4,6]. Although rare, these monogenic forms of diabetes provide a paradigm for understanding and investigating some of the genetic components of more complex forms of diabetes in adults.
The well-defined mode of inheritance of MODY, with a high penetrance and early-onset diabetes, allows the collection of multigenerational pedigrees, making MODY an attractive model for genetic studies. MODY usually develops in thin young adults (usually before 25 years of age; in childhood, adolescence or young adulthood), and is associated with primary insulin-secretion defects [4,5]. The prevalence of MODY is estimated to be less than 1–2% of patients with T2DM, although it could represent as many as 5% of European cases of diabetes [4,25]. MODY is not a single entity, but involves genetic, metabolic and clinical heterogeneity. So far, heterozygous mutations or chromosome rearrangements in seven genes have been identified as responsible for the disease (Table 12.2). These genes encode the enzyme glucokinase (GCK, MODY type 2) [26–28], the transcription factors hepatocyte nuclear factor 4α (HNF-4α//HNF4A, MODY type 1) [29], hepatocyte nuclear factor 1α (HNF-1α/HNF1A, MODY type 3) [30,31], insulin promoter factor 1 (IPF-1, MODY type 4) [32,33], hepatocyte nuclear factor 1β (HNF-1β/TCF2, MODY type 5) [34] and NEUROD1/β2 (MODY type 6) [35], and the preproinsulin (INS) (MODY type 7) [6]. Moreover, additional unknown genes (MODY-X) related to the MODY phenotype remain to be discovered from families in which early-onset diabetes cosegregates with genetic markers outside the known MODY loci [36–38].
Table 12.2 The different subtypes of maturity-onset diabetes of the young ( MODY ).
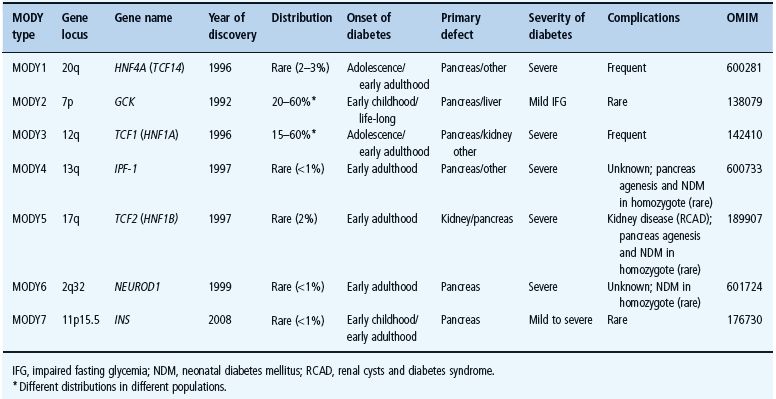
The relative prevalence of the different subtypes of MODY has been shown to vary greatly in studies of British, French, German and Spanish family cohorts [39–42]. These contrasting results may be caused by differences in the genetic background of these populations, or else may reflect, at least partly, ascertainment bias in the recruitment of families. Altogether, mutations in GCK and HNF1A are the cause of the two most prevalent MODY2 and MODY3 subtypes, accounting for around 50–60% of all MODY cases. Mutations in HNF4A and TCF2/Hnf-1β were identified in many dozens of families and the other defects caused by mutations in PDX1 and NEUROD1 are rarer disorders [43,44].
Recent challenging studies have tried to identify a connection between monogenic diabetes genes and common T2DM, that is to test whether common but less severe variants might have a role in the pathogenesis of common multifactorial forms of the disease. Indeed, if major mutations (i.e. causing a substantial functional defect and normally rare or absent in the general population) lead to a highly penetrant form of diabetes, it seems plausible that more subtle genetic changes affecting the structure or expression of the gene product might have a role in determining (minor) susceptibility to T2DM. Our current understanding of genetic variants influencing T2DM supports this hypothesis [3,4,6], but common variants (also referred as single nucleotide polymorphisms [SNPs]) in the known MODY genes seem to contribute very modestly to the common forms of T2DM, as recently assessed in a staged case–control study from multiple clinical samples [45]. In this study, the strongest effects were found for an intronic variant of TCF2/HNF-1β and for the –30 G/A variant of GCK. The –30 G/A polymorphism in the β-cell specific promoter of glucokinase was found to modulate diabetes risk, with the (–30) A-allele being associated with an increased risk) [45]. A similar genetic association was observed with a stronger effect in a French prospective study of a middle-aged general population, along with a significant impact on the modulation of fasting glycemia and insulin secretion HOMA-B index [46]. Furthermore, a meta-analysis of previously reported association results for GCK (–30A) with T2DM showed a modest overall effect on disease risk of 1.08 (P = 0.004) in European populations [45].
Mutations in HNF1α were identified in African-Americans and Japanese subjects with atypical non-autoimmune diabetes with acute onset [47,48]. In the Oji-Cree native Canadian population the G319S mutation in HNF1A, found in approximately 40% of patients with diabetes, accelerates the onset of T2DM by 7 years [49]. Such findings show that HNF1A mutations can be associated with typical adult-onset insulin-resistant obesity-related diabetes in addition to MODY.
A population-based study in Swedish and Finnish cohorts using both in vitro and in vivo experiments has shown that common variants in and upstream of the HNF1A gene influence transcriptional activity and insulin secretion in vivo [50]. Some of these variants are associated with a modestly higher risk of diabetes in subsets of elderly overweight individuals. Mutations in HNF4A and IPF1 genes were also identified in a number of families with late-onset T2DM [51]. IPF-1/Pdx-1 has a dosage-dependent regulatory effect on the expression of β-cell-specific genes and therefore assists in the maintenance of euglycemia. As a consequence, frequent variants in the regulatory sequences controlling IPF-1/Pdx-1 expression in the β-cell, or in genes coding for transcription factors known to regulate IPF-1, could contribute to common T2DM susceptibility. Two independent studies, from the Ashkenazim [52] and Finnish [53] populations, have reported significant associations between common variants adjacent to the HNF4A P2 promoter and T2DM. Interestingly, some of the diabetes-associated variants account for most of the evidence of linkage to chromosome 20q13 reported in these two populations. Consistent with these results, genetic variation near the P2 region of HNF4A is associated with T2DM in other Danish and UK populations, but not in French and other Caucasian populations [54] which argues for genetic heterogeneity in HNF4A variants susceptibility [55].
Recent studies have evaluated the genotype–phenotype correlation between the MODY genes and the common form of T2DM and have reported that several common variants (MAF >0.05) in TCF2/HNF1β may contribute to T2DM risk but with modest effects (allelic odds ratio [OR] <1.25); the strongest effect was found for an intronic variant with corrected P values <0.01 and OR of 1.13 [45]. Independently, a GWA scan performed to search for sequence variants conferring risk of prostate cancer demonstrated with replication from eight case–control groups that two intronic variants located in the first and second intron of TCF2 gene confer protection against T2DM (OR 0.91, P ~ 10–7) in individuals of European, African and Asian descent [56]. Several epidemiologic studies have reported an inverse relationship between T2DM and the risk of prostate cancer, and a recent meta-analysis estimated the relative risk of prostate cancer to be 0.84 (95% CI 0.71–0.92) among patients with diabetes [57]. Previous explanations of this inverse relationship between T2DM and prostate cancer have centered on the impact of the metabolic and hormonal environment in men. The protective effect of the TCF2 SNPs against T2DM is too modest to explain their impact on prostate cancer risk by a consequence of an effect on diabetes. The primary functional impact of TCF2 variants may lie within one or more metabolic or hormonal pathway, and incidentally may modulate the risk of developing prostate cancer and T2DM throughout life.
Genetic variation in the WFS1 gene, which encodes a 890-amino-acid polypeptide named wolframin, a transmembrane protein located in the endoplasmic reticulum (ER), which serves as an calcium channel, not only results in the rare Wolfram syndrome (known as DIDMOAD and characterized by early-onset non-autoimmune diabetes mellitus, diabetes insipidus, optic atrophy and deafness) [58,59] but is also associated with susceptibility to adult T2DM [60]. In a pooled case–control analysis comprising >20 000 individuals, several SNPs in WFS1 (including a non-synonymous SNP, R611H) were shown to modulate diabetes risk (OR ~0.92 for a minor allele frequency of ~40% [60]), with a population attributable fraction of 9%, which could explain 0.3% of the excess familial risk. This study provides further evidence that wolframin has an essential role in the ER stress response in insulin-producing pancreatic β-cells, and contributes to the risk of common T2DM.
Search for T2DM genes in the pre-GWA era
Before the completion of GWA studies in early 2007, numerous focused candidate gene studies, driven by biologic or functional hypotheses, and the hypothesis-free linkage approach, have delivered a small number of validated T2DM susceptibility genes which effects the disease process (both insulin secretion and insulin sensitivity), although contributing at the individual level with a modest risk effect.
Candidate gene approach
The molecular screening of candidate genes to search for genetic variants (either rare when the allele frequency is < 0.01, or common in the population tested) potentially associated with diabetes status (i.e. more frequent in individuals with T2DM) has so far been the most frequently used approach to tackle the genetic determinants of T2DM [61]. There are many reasons why specific genes may be candidates:
- A gene may have a known or presumed biologic function in glucose homeostasis or energy balance in humans.
- It may be implicated in subtypes of diabetes, such as MODY or neonatal diabetes.
- It may be associated with diabetes or associated traits in animal models.
- It may be responsible for an inherited disease that includes diabetes (e.g. mitochondrial cytopathies, Wolfram syndrome).
- The gene products may be differentially expressed in diabetic and normal tissues.
For obvious reasons, the insulin gene was among the first genes to be studied. Apart the more recent identification of rare mutations in the sequence of the preproinsulin gene responsible for neonatal and non-autoimmune early infancy diabetes [5,6], mutations in the coding regions of the insulin gene have been reported to be associated with familial hyperinsulinemia in less than 10 families, but are not consistently associated with T2DM [62]. Mutations in the promoter region, however, could affect the regulation of the insulin gene, leading to absolute or relative hypoinsulinemia. A variant allele of the promoter has been observed in about 5% of African-Americans with T2DM, and shown to be associated with decreased transcriptional activity [63]. An association between T2DM and paternally transmitted class III alleles of the variable number tandem repeat (VNTR) region upstream of the insulin gene (INS-VNTR) was observed in British families [64]. Interestingly, class III alleles (one of the two main classes of INS-VNTR allele length, with 141–209 repeats) were also found to be associated with increased length and weight at birth [65] and with a dominant protection against type 1 diabetes [66] compared with type I alleles.
Key components of the insulin signaling pathways have also been tested. They were at first thought to be important players in the context of the insulin resistance of T2DM. Several of these genes are also expressed in pancreatic β-cells, and several studies from knockout animals have demonstrated that they may also have an important role in the mechanisms of insulin secretion [23,24]. More than 50 different mutations have been found in the coding regions of the insulin receptor gene on chromosome 19p (see Chapter 15) [67]; patients with these mutations seldom present with the common form of T2DM [68], but rather with a syndrome of severe insulin resistance associated with leprechaunism, or with acanthosis nigricans, hirsutism and major hyperinsulinemia [69]. Missense variants in the gene encoding the first substrate for the insulin receptor kinase (IRS1) on chromosome 2q have been detected in several populations [70–73] but an association of these variants with diabetes was not observed in all studies [74,75].
Similarly, an association between polymorphisms of the muscle glycogen synthase gene (GYS1) on chromosome 19q and T2DM has been observed in Finnish [76] and in Japanese [77] subjects, but not in French subjects [78]. Taken together, these results suggest that the IRS1 and GYS1 genes may act in some populations as minor susceptibility genes, which are neither necessary nor sufficient for disease expression, but may nevertheless modulate the phenotype in the patients. This is important in the light of more recent data obtained from combined and meta-analyses conducted by GWA follow-up studies, which report a significant effect of common variants at the IRS1 locus on T2DM risk in European descent populations (unpublished results).
Functionally significant polymorphisms have also been identified in other proteins involved in insulin action, such as the phosphatidylinositol 3 kinase [79] and the ectonucleotide pyrophosphatase/phosphodiesterase 1 enzyme (ENPP1 or PC-1), a transmembrane glycoprotein that downregulates insulin signaling by inhibiting insulin-receptor tyrosine kinase activity [80]. ENPP1 is expressed in multiple tissues including three targets of insulin action: adipose tissue, muscle and liver. A missense variation in its gene ENPP1, in which lysine 121 is replaced by glutamine (K121Q, rs1044498), results in a gain-of-function mutation leading to greater inhibition of the insulin receptor and clinical insulin resistance [81]. Pizutti et al. [82] first identified the association while studying healthy non-obese non-diabetic Sicilian subjects. ENPP1 K121Q is also associated with an earlier onset of T2DM in Europeans and obese children, suggesting that the variant may accelerate disease onset in predisposing individuals. Not all studies have replicated these results, and some studies have reported that high body mass index (BMI) exacerbates, or in some cases is a prerequisite for, the deleterious role of ENPP1 K121Q on insulin resistance-related phenotypes and T2DM [83,84]. Meyre et al. [85] reported an effect of the Q121 variant on the risk of both morbid obesity and hyperglycemia in large samples of French children and adults, as observed in children from Germany [86] and British adults [87]. Given the major role of obesity in deteriorating glucose homeostasis, the different effect of the Q121 variant on BMI in different samples may be, at least partly, responsible for the non-homogeneous results so far reported on the risk of across different studies. A recent large meta-analysis performed on published case–control studies has shown that, although results are not homogeneous across all samples, individuals carrying the variant have an approximately 20% increased risk of T2DM [88].
Another biologic candidate gene that was extensively studied is the peroxisome proliferator-activated receptor γ gene (PPARG), where mutations that severely decrease the transactivation potential were found to cosegregate with extreme insulin resistance, diabetes and hypertension in two families, with autosomal-dominant inheritance [89]. A common amino-acid polymorphism (Pro12Ala) in PPARG has been associated with T2DM; homozygous carriers of the Pro12 allele are more insulin resistant than those having one Ala12 allele and have a 1.25-fold increased risk of developing diabetes [90]. This common polymorphism has a modest, yet extensively replicated effect on the risk of T2DM. There is also evidence for interaction between this polymorphism and the insulin secretion in response to fatty acids [91], and BMI [92]; the protective effect of the alanine allele was lost in subjects with a BMI greater than 35 kg/m2. A widespread Gly482Ser polymorphism of PGC1-α (known as PPARGC1), a transcriptional coactivator of a series of nuclear receptors including PPARG, has been associated with a 1.34 genotype relative risk of T2DM [93]. In this study, a test for interaction with the Pro12Ala variant in PPARG gave no indication for additive effects on diabetes status.
Other genes have been shown to be implicated in the genetic susceptibility to insulin resistance. Although they do not appear to be directly linked or associated with T2DM, they may also modulate the disease expression. A common and widespread polymorphism at codon 905 in the gene encoding the glycogen associated regulatory subunit of protein phosphatase 1 (PP1), which is expressed in skeletal muscle and plays a crucial part in muscle tissue glycogen synthesis and breakdown, has been shown to be associated with insulin resistance and hypersecretion of insulin in Danish subjects with T2DM [94]. A missense mutation in the intestinal fatty acid binding protein 2 (FABP2) gene on chromosome 4q has been found to be associated with increased fatty acid binding, increased fat oxidation and insulin resistance in the Pima Indians of Arizona [95], an ethnic group with the highest reported prevalence of T2DM and insulin resistance in the world. A rare P387L variant in protein tyrosine phosphatase-1B (PTP-1B), a negative regulator for insulin and leptin signaling, has been described to be associated with a 3.7 genotype-relative risk of T2DM in a Danish Caucasian population, resulting in impaired in vitro serine phosphorylation of the PTP-1B protein [96]. In an extensive analysis of the PTPN1 gene locus, Bento et al. [97] found convincing associations between multiple SNPs and T2DM in two independent Caucasian American case–control samples. A modest effect of PTPN1 gene variants on disease risk was observed in the French population, whereas consistent associations with metabolic variables reflecting insulin resistance and dyslipidemia were found for two intronic SNPs, thereby influencing susceptibility to metabolic diseases [98].
Mutations in transcription factors have also been reported to contribute to the genetic risk for T2DM through various mechanisms: dysregulation of target genes involved in glucose or lipid metabolism (HNFs, PPARG, IPF-1, IB1, TIEG2/KLF11), impaired β-cell development and differentiation (IPF-1, NEUROD1/β2, TIEG2/KLF11), and increased β-cell apoptosis (IB1/MAPK8IP1). Deleterious mutations that significantly impair the transactivation activity of these transcription factors can be responsible in some families for monogenic-like forms of diabetes with late age of onset, which may represent an intermediary phenotype between MODY and the most common forms of T2DM. This is the case for the TIEG2/KLF11 gene encoding the Krüppel-like factor 11 (KLF11), an SP1-like pancreas expressed transcription factor that is induced by the transforming growth factor β(TGF-β) and regulates cell growth in the exocrine pancreas. A common polymorphism (Q62R) in KLF11 was reported to be associated with polygenic T2DM developing in adulthood and to affect the function of KLF11 in vitro [99]. Insulin levels were found to be lower in carriers of the minor allele at Q62R [99] but attempts of replication in other populations only found a minor, or no detectable effect of the Q62R common variant on diabetes risk [100]. Sequencing of KLF11 gene in families enriched for early-onset T2DM uncovered two missense mutations which segregated with diabetes in three pedigrees [99], but proof of their causality was only based on in vitro experiments. These findings suggest a role for the TGF-β signaling pathway in pancreatic diseases affecting endocrine islets (diabetes) or exocrine cells (cancer) [101].
Another example is the identification of a mutation in islet brain 1 (IB1, MAPK8IP1) found to be associated with diabetes in one family [102]. IB1 is a homologue of the c-jun amino-terminal kinase interacting protein 1 (JIP-1), which has a role in the modulation of apoptosis [103]. IB1 is also a transactivator of the islet glucose transporter GLUT-2. The mutant IB1 was found to be unable to prevent apoptosis in vitro [102]. It is thus possible that the abnormal function of this mutant IB1 may render the β-cells more susceptible to apoptotic stimuli, thus decreasing β-cell mass. As glucotoxicity and lipotoxicity are known to induce both apoptosis and transcription factor down regulation in pancreatic β-cells, inherited or acquired defects in IB1 activity could have deleterious effects in β-cell function.
Other genes encoding key components of insulin secretion pathways have also been tested as potential candidates for a role in the genetic susceptibility of T2DM. The pancreatic β-cell ATP-sensitive potassium channel (IKATP) has a central role in glucose-induced insulin secretion by linking signals derived from glucose metabolism to cell-membrane depolarization and insulin exocytosis (see Chapter 6) [104]. IKATP is composed of two distinct subunits: an inwardly rectifying ion channel forming the pore (Kir6.2), and a regulatory subunit, which is a sulfonylurea receptor (SUR1) belonging to the ATP-binding cassette (ABC) superfamily [105]; both subunits are expressed in neuroendocrine cells. The KCNJ11 and ABCC8 genes encoding these two subunits are located 4.5 kb apart on human chromosome 11p15.1. Inactivating mutations in each of these genes may result in familial persistent hyperinsulinemic hypoglycemia in infancy, and gain-of-function mutations are responsible of the opposite phenotype of neonatal diabetes (either permanent or transient forms of the disease with distinct distributions of mutations in each gene), demonstrating their role in the regulation of insulin secretion (see Chapter 15) [106]. Studies in various populations with different ethnic backgrounds have provided evidence for associations of SNPs in these genes with T2DM [107–112]. In particular, a common variant, E23K, of KCNJ11/KIR6.2 has now been convincingly associated with an increased risk of T2DM and decreased insulin secretion in glucose-tolerant subjects [113,114]. Large-scale studies and meta-analyses have consistently associated the lysine variant with T2DM, with an OR of 1.15 [115].
Increasing evidence from more recent studies also suggested that inflammatory processes may have a pivotal role in metabolic diseases: prospective studies have shown that high plasma interleukin 6 (IL-6) levels increased T2DM risk [116], but conflicting associations were found between a promoter polymorphism (G-174C) in IL6 and T2DM [117,118]. In a large joint analysis of 21 case–control studies, representing >20 000 participants in one of the largest association studies addressing the role of a candidate gene in T2DM susceptibility, the IL6 promoter variant was found to be associated with a lower risk (OR 0.91, P = 0.037) [119]. In addition, association between T2DM and IL6R-D358A was reported in Danish white people [120], and with TNF G-308A promoter SNP in the Finnish Diabetes Prevention Study [118]. The effects of both IL6 and IL6R variants on developing T2DM risk in interaction with age have been reported in a prospective study of a general French population [46].
Hypothesis-free genome-wide approach and positional cloning of T2DM genes
The second approach used in the years 1995–2005 for identifying genes underlying common polygenic T2DM, the so-called genome-wide familial linkage (GWL) studies, was based on genome-wide scans to detect chromosomal regions showing linkage with diabetes in large collections of nuclear families or sib-pairs. This strategy required no assumptions regarding the function of genes at the susceptibility loci, because it attempted to map genes purely by position. Genotyping of approximately 400 multiallelic markers (short tandem repeats or microsatellites, with a density of ~1 marker/10 cmol) allows identification of polymorphic markers showing strong allele identity by descent in diabetic family members (i.e. allele sharing in sibships is significantly higher than 50%). Once identified, such susceptibility genes for diabetes may then be positionally cloned in the intervals of linkage.
This total genome approach has been used for some time in other multifactorial diseases such as type 1 diabetes [121] and obesity [5]. More than 20 genome scans for T2DM have been completed, involving thousands of pedigrees from different populations and ethnic groups. One of the limitations of the genome-scan approach is the relatively low power of the method, which is unable to detect a weak linkage signal because of the low relative risk for diabetes in siblings (about a three-to fivefold increase in comparison with the general population). Working on very large family collections (>500 sib-pairs), in more homogeneous ethnic groups (e.g. island populations), or in large pedigrees using quantitative intermediary traits instead of the dichotomous diabetes status, has improved the effectiveness of this approach in detecting linkage. Indeed, intermediary phenotypes were shown to have greater heritability than the dichotomous endpoint. To avoid bias brought about by the effect of chronic hyperglycemia on many traits, other strategies have been proposed to analyze the general population [122] or to collect sibships of the offspring of subjects with diabetes who are not yet diabetic but are at high risk of developing the disease [123]. False-positive results are likely to occur. Thus, stringent criteria for linkage (in the magnitude of P <10–5) need to be used to minimize bias brought about by multiple testing. Indeed, a level of statistical significance at the genome-wide level (i.e. giving a less than 5% chance that a linked locus is false) implies a multipoint lod score higher than 3.6 at a given chromosomal region. This value is not a rigid one; each case is different, and sophisticated simulations are needed to provide an empirical P value, and therefore to assess the robustness of the data obtained. Indeed, the validation (or the absence of validation) of a linked locus has strong implications for further research, and careful examination is necessary. For this reason, another criterion for a true locus has been the replication of the findings by others. Although less stringent P values are required (10–3), replication is difficult, because genetic heterogeneity, ascertainment biases and phenotypic differences across studies may mask a confirmatory linkage. Furthermore, genotyping errors have a substantial effect on linkage analyses.
Since 1996, the results of several genome scans in families with diabetes have been published [123]. A summary of the best T2DM linkage replication results is presented in Table 12.3. A locus for T2DM on chromosome 2q (NIDDM1) was initially found in Mexican Americans [14], and it was shown that an interaction between this locus and a locus on chromosome 15 further increases the susceptibility to diabetes in this population [124]. A region of replicated linkage between diabetes and chromosome 1q21-25 was reported in different populations of European origin and in Pima Indians [123]. Linkages with diabetes and with the age at onset of diabetes were found on chromosome 3q27 [125–127], and in a region on chromosome 10q in Mexican American families from San Antonio, Texas [128]. Linkage was found at a locus near TCF1 on chromosome 12q in Finnish T2DM families, characterized by a predominant insulin secretion defect [13]. Evidence for an obesity–diabetes locus on chromosome 11q23-q25 [129] and linkage of several chromosomal regions with prediabetic traits [14] were observed in Pima Indians from Arizona, an ethnic group with a high prevalence of diabetes and obesity. Evidence for the presence of one or more diabetes loci on chromosome 20 was found in different populations [130,131]. In these and other studies, a large number of loci showing only suggestive or weak indication of linkage with diabetes-related traits have also been reported, several of which fall in overlapping regions.
Table 12.3 Genomic locations of best type 2 diabetes linkage replications.
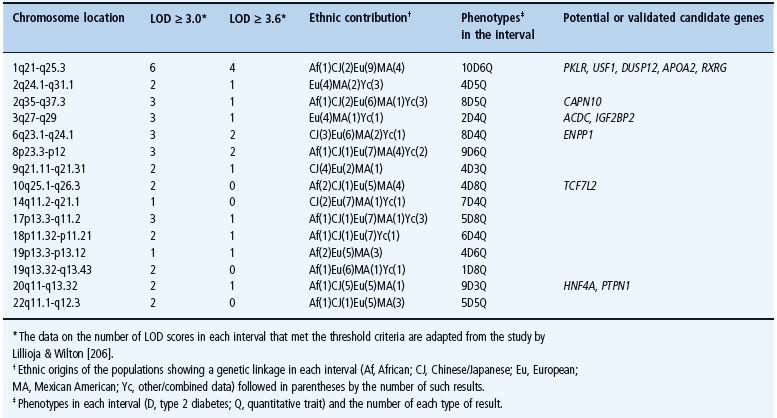
Some concerns have therefore been raised about the heterogeneity and reliability of genetic data in multifactorial diseases in general (e.g. the lack of replication), and in T2DM in particular. A new series of genome scans published in 2000 and later [125–127], however, has surprisingly shown that results obtained from T2DM families may be reproducible, despite differences in ethnicity and in environmental factors. It is likely that progress both in automated genotyping and statistical analyses of larger sample sets have improved the quality of the data. A meta-analysis of the data obtained from all European studies has confirmed findings on 1q, 2p and 12q, and also revealed a new locus on 17p linked to the metabolic syndrome in the US population [132,133]. These data provide a strong basis for believing that some important genetic contributors to common T2DM exist.
Although genome-wide scans have mapped T2DM loci, each hit represents 10–20 million nucleotides. The challenge was to identify the diabetes-related genes within these intervals, knowing that hundreds of genes may be present. To identify the true etiologic gene variants associated with an enhanced risk for T2DM, one first positional cloning approach was to refine the chromosomal regions of linkage using a dense map of biallelic SNPs as markers for the detection of linkage disequilibrium (LD) mapping [134–136]. DNA polymorphisms located away from the true functional variant may be associated with the disease, or with the variation of a diabetes-associated trait. The strength of LD is quite variable within the genome, ranging from 10 to 300 kb or more. It has been postulated that working in a so-called “isolated” population should significantly enhance results with LD, but even research in Finns or in Icelanders may not resolve the problems [137].
The identification of the NIDDM1/CAPN10 gene (coding for calpain-10, a non-lysosomal cysteine protease) on 2q37 confirmed that LD mapping may be a successful strategy for unravelling other polygenic diseases [138]. This work, however, also showed the complexity of the search, because in this case an intronic polymorphism (UCSNP-43) was associated with T2DM in Mexican Americans. In fact, three non-coding polymorphisms, including SNP-43, SNP-44 and SNP-63, have been identified as defining an at-risk haplotype [138]. In other ethnic groups, such as French Caucasians, the rarity of this high-risk haplotype made it difficult to achieve a definite answer concerning the role of calpain-10 in T2DM. Nonetheless, recent pooled and meta-analyses as well as the LD and haplotype diversity studies suggest a role for genetic variation in CAPN10 affecting risk of T2DM in Europeans [139]. CAPN10 was thus the first diabetes gene to be identified through a genome-wide scan approach. Many investigators, but not all, have subsequently found associations between CAPN10 polymorphism(s) and T2DM as well as insulin action, insulin secretion, aspects of adipocyte biology and microvascular functions. This has not always been with the same SNP or haplotype or the same phenotype, suggesting that there might be more than one disease-associated CAPN10 variant and that these might vary between ethnic groups and the phenotype under study. Indeed, both genetic and functional data indicated that calpain-10 has an important role in insulin resistance and intermediate phenotypes, including those associated with the adipocyte [140]. In this regard, emerging evidence would suggest that calpain-10 facilitates GLUT-4 translocation and acts in the reorganization of the cytoskeleton. Interestingly, calpain-10 is also an important molecule in the β-cell. It is likely to be a determinant of fuel sensing and insulin exocytosis, with actions at the mitochondria and plasma membrane, respectively, and also of pancreatic β-cell apoptosis [141].
Only a few other chromosomal regions have shown highly significant lod scores in several populations. The best example is probably the chromosome 1q21-25 region (in particular the 30 Mb stretch adjacent to the centromere), which ranks alongside the regions on chromosomes 10 and 20 as amongst the strongest in terms of the replicated evidence for GWL to T2DM. Linkage has been reported in samples of European (UK, French, Amish, Utah), East Asian (Chinese, Hong Kong) and Native American (Pima) origin (summarized in [123]). The homologous region has also emerged as a diabetes-susceptibility locus from mapping efforts in several well-characterized rodent models [142]. The human region concerned is gene-rich and contains a disproportionate share of excellent biologic candidates, and genetic variation in a number of them has already been investigated with details in the populations showing 1q21-linkage (e.g. the genes USF1 encoding the upstream stimulatory factor 1, LMNA1 encoding Lamin A/C, PBX1 encoding a TALE-homeodomain transcription factor that has a role in pancreatic development and is an important cofactor of Pdx1, the “master regulator” of pancreatic development and function, ATF6 encoding activating transcription factor 6, or DUSP12 encoding the glucokinase-associated, dual-specificity phosphatase 12). These studies have been conducted by an international consortium, which represent a coordinated effort by the groups with the strongest evidence for 1q linkage, by using a high-density, custom LD mapping approach applied to a well-powered set of multi-ethnic samples to identify variants causal for that signal [143]. Detection of low-frequency susceptibility variants also requires new approaches and the future plans of the 1q consortium include deep resequencing of the 1q region of interest, focusing at least initially on exons and conserved sequence.
Linkage with chromosome 20 has been also found in several ethnic groups, from Finns to Japanese subjects [147], but the linked region is rather large, raising the issue of whether there may be several susceptibility genes. While common variants upstream of HNF4A that maps to chromosome 20q13 have been reported to explain the linkage signals seen in Finns and Ashkenazim [144,145], these associations have proved difficult to replicate in many other populations [146].
Loci overlapping between T2DM, obesity and the metabolic syndrome have appeared on chromosome 2p [125,147] and on chromosome 3q27 [125,126,127]. Several candidate genes map to the 3q27 region, including the APM1/ACDC gene encoding the differentiated adipocyte-secreted protein, adipocyte complement-related protein 30 (Acrp30/adiponectin), which is an adipokine abundantly present in plasma. The purified C-terminal domain of adiponectin has been reported to protect mice on a high-fat diet from obesity, and to rescue obese or lipoatrophic mice models from severe insulin resistance, by decreasing levels of plasma free fatty acids (FFAs) and enhancing lipid oxidization in muscle [148]. Moreover, plasma levels of adiponectin have been shown to be decreased in obese subjects with diabetes and to correlate with insulin sensitivity [149], which makes ACDC/ACRP30 an attractive candidate gene for fat-induced metabolic syndrome and T2DM.
Both common and rare variants of ACDC/ACRP30 were found to be associated with T2DM in numerous obese populations, but do not contribute to the 3q27 linkage in the French families, suggesting that other genes may be involved. Common variants in and upstream of EIF4A2 (encoding the eukaryotic translation initiation factor 4 α2) have been shown to contribute to both age of onset of diabetes and linkage in the French families studied [150].
One notable exception is the identification of TCF7L2 as a T2DM gene with the largest risk effect of intronic variants of any diabetes gene identified to date. In early 2006, common intronic variants of the TCF7L2 gene encoding the transcription factor 7-like 2 (TCF7L2) were identified to be strongly associated with T2DM risk in the Icelandic population by DeCODE Genetics (Reykjavik, Iceland) during a follow-up positional cloning study from a modest linkage signal on chromosome 10q [151]; but it turned out that despite not explaining this linkage signal, multiple intronic SNPs also showed strong association in US and Danish samples [151]. This initial study was rapidly followed by widespread replications in different ethnic backgrounds from white Europeans, West Africans, Mexican Americans, Indian and Japanese populations [152], confirming that TCF7L2 variants have the largest risk effect of any of the known T2DM susceptibility loci. As shown in a meta-analysis from >20 studies in diverse ethnicities, the estimated risk of rs7903146 T allele (the most T2DM-associated variant) was 1.46 (P = 5.4 × 10–140) [153].
At the individual level, carrying one TCF7L2 risk allele may increase diabetes risk by 40%–60% [152]. As a marked variation in TCF7L2 rs7903146 T allele frequency exists between individuals from different ethnic groups, this implies a variable impact in the respective general populations. In European general populations, rs7903146 T allele has been estimated to contribute to 10–25% of all cases of diabetes in lean individuals [152]. In Eastern Asians, the population attributable risk fraction was assessed at 18.7% for other specific risk alleles [154]. The rs7903146 variant has been shown to influence progression to diabetes (with a hazard ratio of 1.81 for the homozygote TT group in the participants from the Diabetes Prevention Program) [155].
In the French and UK populations, rs7903146 T allele was associated with lower BMI in individuals with T2DM and with a higher risk for T2DM in non-obese subjects [152], but it is not a risk factor for obesity in European populations, whereas its effect on T2DM risk was found to be modulated by adiposity [156]. Watanabe et al. [157] suggested that an interaction between TCF7L2 and adiposity may be caused by a better β-cell compensation in obese individuals.
TCF7L2 is a member of the T-cell specific high mobility group (HMG) box containing family of transcription factors which, upon binding β-catenin, transduces signals generated by Wnt receptors at the cell surface to modify expression of multiple genes, many of which are associated with the cell cycle. Mutations in TCF7L2 are also implicated in certain types of cancer [158], but rare TCF7L2 mutations are not contributing to monogenic forms of diabetes such as MODY and neonatal diabetes [159]. One mechanism whereby TCF7L2 variants increase diabetes risk might involve an impairment of incretin effects (i.e. GLP-1) on islet functions [160,161]. Indeed, variants of TCF7L2 specifically impair GLP-1-induced insulin secretion [162], and the stimulation of β-cell proliferation by GLP-1 is TCF7L2 dependent [163]. TCF7L2 is also required to maintain glucose-stimulated insulin secretion and β-cell survival because its deletion in human islets reduces insulin secretion and increased apoptosis [164]. Furthermore, the overexpression of TCF7L2 protects β-cells from glucose and cytokine-induced apoptosis [164]. Clinical studies have demonstrated that the at-risk SNP is associated with defects in β-cell function and insulin release. The study by Lyssenko et al. [161] suggested that pancreatic islets from subjects with T2DM bearing the at-risk TT genotype have increased TCF7L2 RNA levels, indicating that excessive TCF7L2 expression may be associated with the reported insulin secretory defects. Expression studies carried out by Elbein et al. [165] on adipose tissue and muscle, however, showed that TCF7L2 expression was not altered by genotype and did not correlate with insulin sensitivity or BMI. As the most associated SNP rs7903146 is found in an intronic region, a potential mechanism through which it may confer an increased risk of diabetes is by altering TCF7L2 expression levels, but the functional mechanisms through which TCF7L2 modulates glucose sensing and insulin release in pancreatic β-cells remain to be clarified. A recent study showed that TCF7L2 silencing in primary mouse islets as well as in clonal MIN6 and INS-1(832/13) cells decreases both basal and glucose-stimulated insulin secretion [166]. TCF7L2 silencing also abolished the stimulatory effects of GLP-1 on secretion, whilst having little impact on glucose-induced changes in intracellular ATP or free Ca2+ concentrations. The study by da Silva Xavier et al. [166] showed that TCF7L2 controls the expression of genes involved in insulin granule fusion at the plasma membrane (notably syntaxin 1A and Munc 18-1 which may be direct targets for regulation by TCF7L2). Thus, defective insulin exocytosis may underlie increased diabetes incidence in carriers of the at-risk TCF7L2 alleles.
The era of GWA studies improved knowledge of T2DM susceptibility genes
New discoveries in the genetic etiology of T2DM
Important advances in T2DM genetics have been made with the completion of GWA studies based on HapMap-selected common SNPs. This has become reality with the outstanding breakthroughs made in the knowledge and assessment of human genome variations, their mapping and their links with the genetic background of common diseases [167], and in the development and accessibility to very high throughput genotyping techniques based on microarray technology and to biostatistical tools for large cohort data analyses.
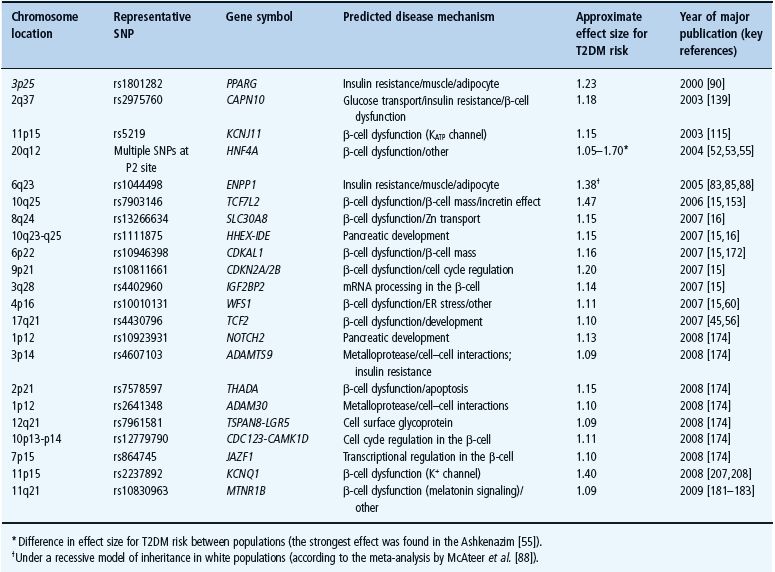
Within only few months of 2007, five independent GWA screens for T2DM, based on the Illumina 300K or Affymetrix 500K genotyping arrays both capturing about 80% of common variants present in the human genome of Caucasian individuals, identified dozens of potential candidates; at least 15 new loci emerged as being most consistently associated with risk of T2DM across multiple studies (Table 12.4). These include variants within or near SLC30A8, HHEX, CDKAL1, CDKN2A/2B, IGF2BP2 and JAZF1 [15]. Three of the associated loci correspond to genes that had already been implicated in T2DM (TCF7L2, KCNJ11, PPARG). These studies also confirmed that TCF7L2 was the highest genetic risk factor for T2DM in populations of European descent. The next strongest SNP is in a 33-kb LD block within the coding region of SLC30A8 encoding a zinc membrane transporter (ZnT8) which is highly expressed in pancreatic islets [168]. Interestingly, the overexpression of ZnT8 in the HeLa cell line is associated with an accumulation of zinc in intracellular vesicles [168]. In the INS-1E β-cell line, which overexpresses ZnT8, the intracellular zinc level is increased and the insulin secretion is stimulated [169]. Furthermore, overexpressing ZnT8 does not sensitize the cells to toxic doses of zinc but confers a resistance to apoptosis after zinc depletion [169]. The non-synonymous polymorphism showing an increased risk of T2DM (OR per risk allele: 1.18) corresponds to an arginine to tryptophan substitution at position 325, and the at-risk C-allele is associated with impaired β-cell function in vivo. An impaired transporter function may decrease the amount of zinc available for co-crystallization with insulin in the secretory vesicles of β-cells. SLC30A8/ZnT-8 has also recently been identified as an autoantigen in human type 1 diabetes [170].
Table 12.4 Validated type 2 diabetes susceptibility genes in the post genome-wide association era.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


