- Glycemic control is a fundamental part of the management of type 2 diabetes mellitus. Adequate glycemic control is necessary to address acute symptoms and to prevent, defer or reduce the severity of chronic microvascular and macrovascular complications.
- Treating type 2 diabetes is complicated by the multivariable and progressive natural history of the disease. Insulin resistance, a progressive decline in β-cell function, defects of other gluco-regulatory hormones and nutrient metabolism give rise to a continually changing presentation of the disease that requires therapy to be adjusted accordingly. Patients are often overweight or obese, exhibit substantial co-morbidity and elevated cardiovascular risk, and receive many other medications which further complicate treatment.
- Care plans and treatment programs should be tailored to fit the prevailing circumstances of the patient. Lifestyle management (diet and exercise) should be emphasized from the time of diagnosis and reinforced thereafter. Drug treatment should be undertaken promptly if lifestyle intervention does not achieve adequate glycemic control.
- Choice of drug therapy should ideally address underlying pathophysiology, but any safe means of restraining the escalating hyperglycemia may be appropriate. Combinations of differently acting agents are frequently required to provide additive efficacy, and single tablet, fixed dose combinations are available to facilitate combination therapy. Contraindications and precautions associated with each component must be respected.
- The biguanide metformin is often selected as initial oral antidiabetic drug therapy. It counters insulin resistance and lowers blood glucose through several insulin-dependent and independent mechanisms, notably reducing hepatic glucose production and also increasing glucose uptake by skeletal muscle. It does not stimulate insulin secretion, carries a low risk of frank hypoglycemia and does not cause weight gain. Metformin also exerts several potentially beneficial effects on cardiovascular risk factors independently of glycemic control, with evidence of improved long-term cardiovascular outcomes. Metformin may be conveniently combined with other classes of antidiabetic drugs. Gastrointestinal side effects including diarrhoea limit the use of metformin. The rare but serious adverse effect of lactic acidosis excludes the use of the drug in patients with significant renal insufficiency, significant liver disease or any condition predisposing to hypoxia or hypoperfusion including cardiac or respiratory failure.
- Sulfonylureas (e.g. gliclazide, glimepiride, glibenclamide/glyburide, glipizide) act on the pancreatic β-cells to stimulate insulin secretion. They bind to the transmembranal complex of sulfonylurea receptors SUR1 with ATP-sensitive Kir6.2 potassium efflux channels. This closes the channels, depolarizes the membrane, opens voltage-dependent calcium channels and raises intracellular free calcium concentrations. This in turn activates proteins regulating insulin secretion. The efficacy of sulfonylureas depends on adequate remaining function of the β-cells. Hypoglycemia is the most serious adverse effect, particularly with longer acting sulfonylureas and in the elderly. Caution with hepatic and/or renal insufficiency is warranted in accordance with the metabolism and elimination of individual preparations, and interactions with other protein-bound drugs can occur.
- Meglitinides (repaglinide and nateglinide), also known as prandial insulin releasers, are rapid and short-acting insulin secretagogues taken before meals to boost insulin levels during digestion, thereby reducing prandial hyperglycemia and decreasing risk of interprandial hypoglycemia. They act in a similar manner to sulfonylureas by binding to a “benzamido” site on the SUR1–Kir6.2 complex. They are conveniently used in combination with an agent that reduces insulin resistance.
- Thiazolidinediones (pioglitazone and rosiglitazone) produce a slow-onset glucose-lowering effect, attributed mainly to increased insulin sensitivity. They alter the expression of certain insulin-sensitive genes by stimulating the peroxisome proliferator-activated receptor γ, increasing adipogenesis, and rebalancing the glucose–fatty acid (Randle) cycle. Thiazolidinediones can be used as monotherapy or in combination with other classes of antidiabetic agents. They have low risk of hypoglycemia but often cause weight gain. The potential for fliud retention and an attendant risk of congestive heart failure should be borne in mind, especially in combination with insulin. Thiazolidinediones are not recommended for individuals at high risk of cardiac disease or women with reduced bone density.
- Gliptins (sitagliptin, vildagliptin and saxagliptin) act predominantly as prandial insulin secretagogues by raising the circulating concentrations of endogenous incretin hormones, notably glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide. Incretin hormones are released from the intestine during a meal and potentiate nutrient-stimulated insulin secretion, but they are rapidly degraded by the enzyme dipeptidyl peptidase 4: gliptins inhibit this enzyme. Gliptins are weight neutral and, as monotherapy, they carry low risk of interprandial hypoglycemia. They are often used in combination with metformin or a thiazolidinedione.
- α-Glucosidase inhibitors (e.g. acarbose) slow the digestion of carbohydrates by competitive inhibition of intestinal α-glucosidase enzymes. This delays glucose absorption and reduces post-prandial glucose excursions without stimulating insulin secretion. These agents must be used in conjunction with meals rich in digestible complex carbohydrate. They do not cause weight gain or hypoglycemia as monotherapy and can be used alongside any other antidiabetic agents.
- In advanced stages of type 2 diabetes, β-cell function is often too severely compromised to support the continued use of oral agents alone. Insulin therapy should be initiated, continuing one or more oral agents where appropriate.
Introduction
Appropriate glycemic control is a fundamental pillar in the management of type 2 diabetes mellitus (T2DM). It is required to prevent and relieve acute symptoms and complications of hyperglycemia; prevent, defer and reduce the severity of microvascular complications; and afford some benefits against macrovascular complications (Table 29.1) (see Chapters 20) [1,2]. Treatment of the hyperglycemia is essential to an individualized care plan that takes account of coexistent diseases and personal circumstances, offers suitable advice on lifestyle and diet, includes other measures to address modifiable cardiovascular risk, selects realistic targets, and facilitates patient education and empowerment, as considered in detail in Part 5. This chapter focuses on the role of oral blood glucose-lowering agents (other antidiabetic therapies are addressed in Chapters 27 and 30) in the treatment of T2DM) [2–5].
Table 29.1 Aims of appropriate glycemic control in type 2 diabetes. Adapted from Wallace & Matthews [1].
| Purpose | Complications |
| Prevent acute symptoms of hyperglycemia | Dehydration, thirst, polyuria, blurred vision, increased infections |
| Prevent acute complications | Hyperosmolar non-ketotic state |
| Prevent, defer or reduce severity of chronic vascular complications | Microvascular: retinopathy, nephropathy, neuropathy |
| Macrovascular: coronary, cerebrovascular, peripheral vascular |
Pathophysiologic considerations
The interdependent multiplicity of genetic and environmental factors underlying T2DM (see Chapters 12 and 14) give rise to a highly heterogeneous and progressive natural history [2,6,7]. The pathophysiology typically involves defects of insulin secretion and insulin action [8]. Obesity, especially visceral adiposity, and abnormalities of glucagon secretion commonly contribute to the disease process, while cellular disturbances of nutrient metabolism participate both as causes and consequences of glucotoxicity and lipotoxicity [9–11]. An ideal approach to therapy might therefore address the basic endocrine defects, but any other safe means of ameliorating the hyperglycemia and attendant biochemical disruptions should provide clinical benefits.
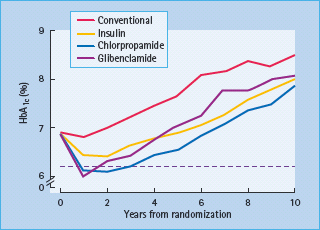
The progressive nature of T2DM was well-illustrated by the UK Prospective Diabetes Study (UKPDS), a randomized trial of 5102 patients with newly diagnosed T2DM followed for a median of 10 years while receiving either “conventional” (diet) therapy or “intensive” therapy with various oral antidiabetic agents or insulin (Figure 29.1). Note that insulin was introduced earlier than is usual in clinical practice, and insulin was also used as necessary when oral agents were deemed inadequate. Although glycemic control (illustrated by the HbA1c level) deteriorated with time irrespective of the treatment, the improvement in glycemic control afforded by intensive therapy (median HbA1c reduced by 0.9% [10 mmol/mol]) was associated with a 12% reduction in overall diabetes-related endpoints and a 25% reduction in microvascular endpoints [12]. An epidemiologic analysis showed that benefits of intensive therapy continued to accrue until glucose levels were returned to the normal range [13]. Moreover, the benefits of earlier “intensive” control were continued during an unrandomized post-trial follow-up (median 8.5 years) during which glycemic differences between the former groups were not maintained [14]. This illustrates the glycemic “legacy” effect in which early intensive glycemic control confers an extended reduction in complications, even when control deteriorates at later stages in the disease process.
Figure 29.1 The UK Prospective Diabetes Study (UKPDS) shows the progressive rise in HbA1c occurring with time in groups receiving “conventional” (diet) therapy and “intensive“ therapy with various antidiabetic drugs (two sulfonylureas – chlorpropamide and glibenclamide – and insulin). Data from Wallace & Matthews [1] and UKPDS [12].
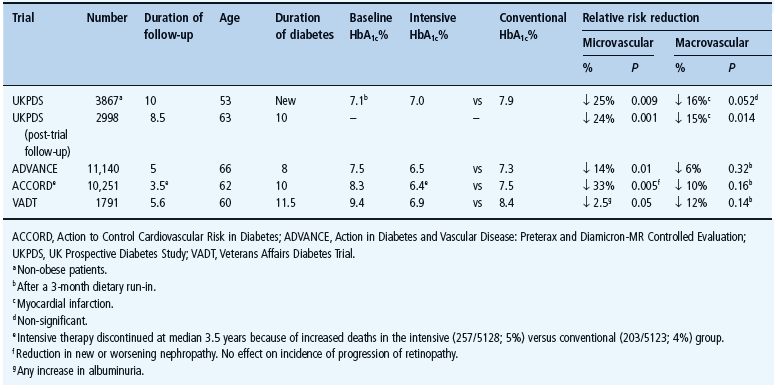
Other large randomized trials [15–17] have confirmed fewer microvascular complications amongst those receiving more intensive glycemic management (Table 29.2). One such study, the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study, noted increased mortality during highly intensified (5% mortality, 257/5128) versus standard (4% mortality, 205/5123) glycemic management. This may be ascribed to unique features of the trial design outside of normal practice in an aging population at high cardiovascular risk with a long previous duration of inadequately controlled diabetes given extensive medications and experiencing high rates of hypoglycemia. Indeed, an acceptable HbA1c value does not preclude excessive daily fluctuations in glycemia with hyperglycemic excursions and hypoglycemic troughs, the latter often unrecognized nocturnally (see Chapter 33). Survival of a myocardial event appears to be reduced by hypoglycemia as well as hyperglycemia [18].
Table 29.2 Trials comparing intensive with standard (conventional) glycemic control in type 2 diabetes.
Guidelines and a lgorithms
Factors to consider when selecting a glycemic target for a particular patient are deliberated in detail in Chapter 20, but it is pertinent to reiterate here that the general principle is to safely return glycemia as close to normal as practicable, avoiding hypoglycemia, minimizing potential drug interactions, and observing other necessary cautions and contraindications. An individualized approach is recommended. Current treatment algorithms [19–25] provide a framework for initiating and intensifying therapy, but clinical judgment should be applied to harmonize this with patient circumstances. Thus, a younger, newly diagnosed individual without co-morbidity who is responsive to therapy might be expected to meet a more rigorous target, while an elderly infirm individual with co-morbidity and a long history of problems with diabetes control may require more flexiblity. Management of hyperglycemia should always be part of a comprehensive management program to address coexistent disease and modifiable cardiovascuar risk factors.
It is emphasized that diet, exercise and other lifestyle measures should be introduced at diagnosis and reinforced at every appropriate opportunity thereafter. These measures can provide valuable blood glucose lowering efficacy and may initially enable the desired glycemic target to be achieved (see Chapters 22 and 23); however, even when lifestyle advice is successfully implemented, the progressive natural history of the disease dictates that the majority of patients will later require pharmacologic therapy, and this should be introduced promply if the glycemic target is not met or not maintained.
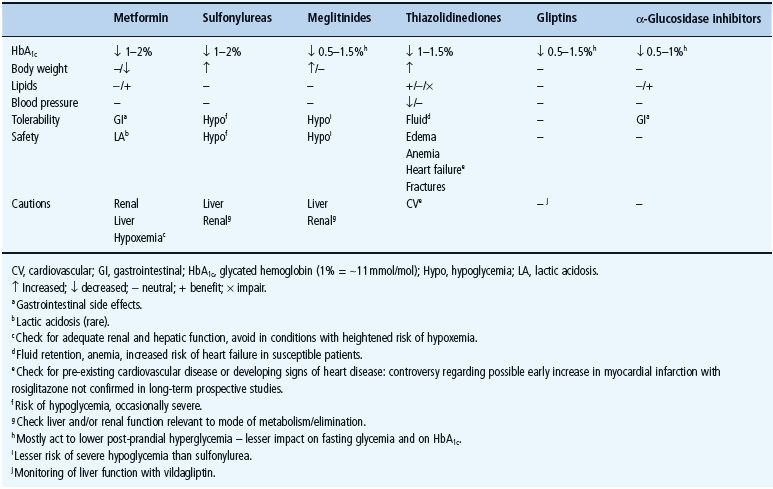
The main classes of oral antidiabetic drugs and their principal modes of action are listed in Table 29.3. The main tissues through which they exert their glucose-lowering effects are illustrated in Figure 29.2, and the main cautions and contraindications associated with oral antidiabetic agents are listed in Table 29.4. Although there are several different classes from which to choose, many dilemmas continue to impinge on both strategy and individualization of treatment. For example, an increase in fasting glycemia usually accounts for the majority of the hyperglycemia in T2DM: ideally therefore, this should be adequately addressed during therapy [26]. It is also pertinent to note the link between post-prandial hyperglycemic excursions and cardiovascular risk, which mandates the need to also address this component of the hyperglycemic day profile [27]. Additionally, consideration should be given to the improvements in glycemic control that can be achieved through the treatment of obesity (see Chapter 14) [28]. By the time of diagnosis, insulin resistance is usually well established and does not usually progress with extended duration of the disease [8]. Nevertheless, the association between insulin resistance and cardiovascular risk warrants the amelioration of insulin resistance as a valued therapeutic strategy. The ongoing deterioration in glycemic control after diagnosis is largely attributed to a further progressive decline in β-cell function [8]. Thus, preserving β-cell function and mass are important considerations in maintaining long-term glycemic control. If β-cell function deteriorates beyond the capacity of oral agents to provide adequate glycemic control, then the introduction of insulin should not be delayed. Incorporating some or all of the above into the treatment process is inevitably a challenge, and the need to explore suitable combinations of therapies to accommodate the changing status of the disease is now accepted practice.
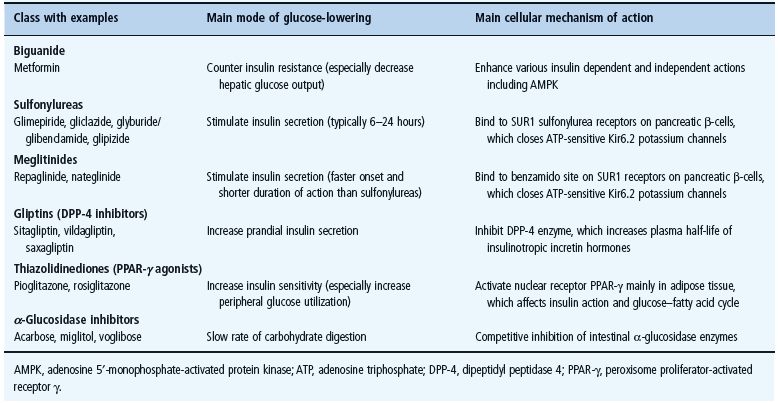
Figure 29.2 Main tissues through which oral antidiabetic agents exert their glucose-lowering effects.
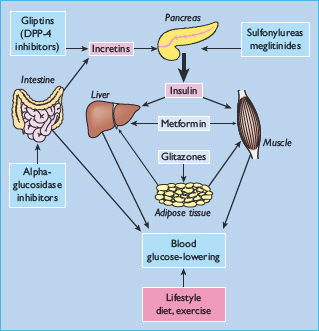
Table 29.4 General features of oral antidiabetic treatments for type 2 diabetes including the main cautions and contraindications.
The increasing prevalence of T2DM amongst younger adults and pre-adults adds an extra long-term dimension to risk–benefit considerations. While initial adequate intervention remains paramount, there is limited experience with oral antidiabetic agents in children and adolescents; metformin has been used safely in controlled pediatric practice from 10 years of age, and sulfonylureas have been used in pediatric presentations of certain forms of maturity-onset diabetes of the young (MODY). Treating T2DM in women who are of childbearing age carries the risk of unplanned pregnancy while receiving oral antidiabetic agents. Treatment with these agents at the time of conception and during the first trimester has not been shown to have any adverse effects on mother or fetus, and judicious use of metformin has been shown to reduce miscarriage and gestational diabetes. Insulin remains the preferred antidiabetic medication in pregnancy, however, having a substantial evidence base for safety and flexibility. A paucity of evidence contraindicates thiazolidinediones and gliptins during pregnancy and lactation.
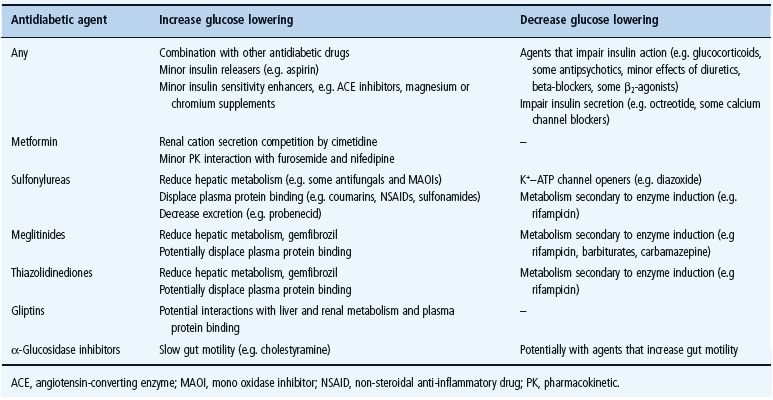
Elderly patients are more vulnerable to most of the cautions and contraindications to glucose-lowering drugs, and deteriorations in their pathophysiologic status can occur rapidly, necessitating more frequent monitoring (see Chapter 54). Hypoglycemia is a particular concern in this age group. While safety must be judged on an individual drug – patient basis, it is noteworthy that several common medications can impair glycemic control (e.g. glucocorticoids, certain antipsychotics, diuretics and beta-blockers), while others may have their own minor glucose-lowering effect (e.g. aspirin, some angiotensin-converting enzyme [ACE] inhibitors and mineral supplements) (see Chapter 26). The most frequent interactions with glucose-lowering drugs are summarized in Table 29.5.
Table 29.5 Drug interactions with oral antidiabetic agents that may affect their glucose-lowering effects.
Terminology within the field of antidiabetic agents may simplify the usage of the different agents. Hypoglycemic agents have the capacity to lower blood glucose below normal to the extent of frank hypoglycemia (e.g. sulfonylureas and insulin). Antihyperglycemic agents can reduce hyperglycemia, but when acting alone they do not have the capability to lower blood glucose below normoglycemia to the extent of frank hypoglycema (e.g. metformin, thiazolidinediones, incretins, gliptins, α-glucosidase inhibitors).
Biguanides
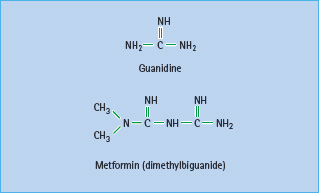
Metformin (dimethylbiguanide) is the only biguanide currently used in most countries (Figure 29.3). The history of biguanides stems from a guanidine-rich herb Galega officinalis (goat’s rue or French lilac) that was used as a traditional treatment for diabetes in Europe [29]. Guanidine has a glucose-lowering effect, and several guanidine derivatives were adopted for the treatment of diabetes in the 1920s [30]. These agents all but disappeared as insulin became available, but three biguanides – metformin, phenformin and buformin – were introduced in the late 1950s. Phenformin and buformin were withdrawn in many countries in the late 1970s because of a high incidence of lactic acidosis. Metformin remained and was introduced into the USA in 1995 [31], and this has since become the most prescribed antidiabetic agent worldwide [32].
Mode of action
Metformin exerts a range of actions that counter insulin resistance and lower blood glucose: the drug also offers some protection against vascular complications independently of its antihyperglycemic effect (Table 29.6) [33,34]. At the cellular level, metformin exerts insulin-dependent and independent effects on glucose metabolism (Figure 29.4). For example, metformin modestly improves insulin sensitivity probably via post-receptor signaling pathways for insulin, and it also appears to influence nutrient metabolism and energy production independently of insulin via activation of adenosine 5 ′-monophosphateactivated protein kinase (AMPK) [35].
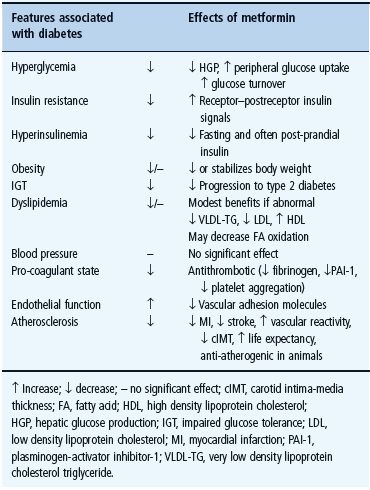
Figure 29.4 Multiple cellular action mechanisms of metformin involve insulin dependent and independent effects. For example, metformin can improve insulin sensitivity via effects on insulin receptor signaling and post-receptor signaling pathways of insulin action. Metformin also appears to influence cellular nutrient metabolism and energy production independently of insulin via activation of adenosine 5′-monophosphate (AMP) activated protein kinase (AMPK). Oct1, organic cation transporter 1; LKB1 protein kinase.
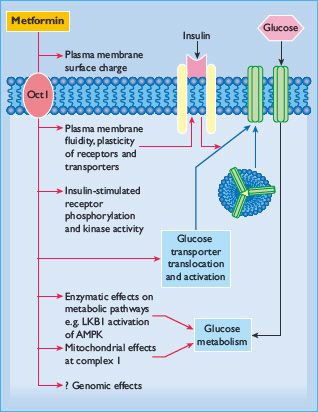
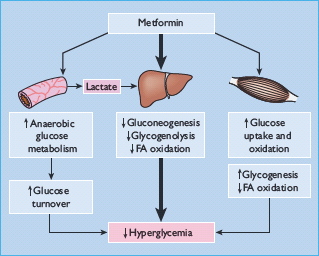
The glucose-lowering efficacy of metformin requires a presence of at least some insulin because metformin does not mimic or activate the genomic effects of insulin. Also, metformin does not stimulate insulin release; its main glucose-lowering effect appears to be a reduction of hepatic glucose production, but not sufficiently to cause frank hypoglycemia when used as mono-therapy. Metformin reduces gluconeogenesis by increasing hepatic insulin sensitivity and by decreasing hepatic extraction of some gluconeogenic substrates such as lactate (Figure 29.5). It also decreases hepatic glycogenolysis. Metformin can enhance insulin-stimulated glucose uptake in skeletal muscle by increasing translocation of insulin-sensitive glucose transporters (GLUT-4) into the cell membrane and increasing activity of glycogen synthase which promotes glycogen synthesis. Independently of insulin, metformin can suppress the oxidation of fatty acids and may reduce triglyceride levels in patients with hypertriglyceridemia. This restricts the supply of energy for hepatic gluconeogenesis and rebalances the glucose–fatty acid (Randle) cycle, allowing greater use of glucose relative to fatty acids as a cellular source of energy. Accumulation of a high cellular concentration of metformin (e.g. in the intestine) can suppress the mitochondrial respiratory chain at complex 1. This may contribute to the blood glucose-lowering effect of the drug, and help to prevent body weight gain by increasing glucose–lactate turnover.
Pharmacokinetics
Metformin is rapidly but incompletely absorbed, shows little binding to plasma proteins and is not metabolized, so it does not interfere with co-administered drugs. Metformin is widely distributed at concentrations similar to plasma (about 10–5 mol/L), but much higher concentrations are retained in the walls of the gastrointestinal tract. The plasma half-life (t1/2) is about 6 hours with elimination of unchanged drug in the urine, mostly within 12 hours [36]. Although renal clearance is achieved more by tubular secretion than glomerular filtration, metformin is contraindicated for patients with significant impairment of glomerular filtration. Cimetidine is the only drug known to compete for clearance sufficiently to cause a clinically significant increase plasma metformin concentrations.
Indications and contraindications
Because metformin does not cause weight gain it is often preferred for overweight and obese people with T2DM, although it shows similar antihyperglycemic efficacy in normal weight patients [37]. To preclude drug accumulation, patient suitability should be considered very carefully if there is any evidence of impaired renal function (e.g. if serum creatinine >130μmol/L or creatinine clearance <60 mL/min or estimated glomerular filtration rate [eGFR] <45mL/min/1.73m2). Further contraindications include significant cardiac or respiratory insufficiency, or any other condition predisposing to hypoxia or reduced tissue perfusion (e.g. hypotension, septicemia), as well as significant liver disease, alcohol abuse or a history of metabolic acidosis. Because the potential for acute deterioration in renal, cardiopulmonary and hepatic function should be taken into account, it is not practicable to identify precise cutoffs for starting or stopping metformin therapy. With this in mind, metformin can be used in the elderly providing renal insufficiency and other exclusions are not present. Ovulation can resume in women with anovulatory polycystic ovarian syndrome (PCOS), which is an unlicensed application of the drug in the absence of diabetes [38].
A standard (so-called immediate release [IR]) tablet or liquid formulation of metformin should be taken with meals or immediately before meals to minimize possible gastrointestinal side effects. Start with 500 or 850 mg once daily, or 500 mg twice daily (divided between the morning and evening meals). The dosage is increased slowly – one tablet at a time – at intervals of about 1–2 weeks until the target level of blood glucose control is attained. If the target is not attained and an additional dose produces no further improvement, return to the previous dose. In the case of monotherapy, consider combination therapy by adding in another agent (e.g. an insulin secretagogue or thiazolidinedione). The maximal effective dosage of metformin is about 2000 mg/ day, taken in divided doses with meals, and the maximum is 2550 or 3000 mg/day in different countries.
Slow-release formulations (XR/SR/ER) of metformin are available in most countries; they can be taken once daily in the morning, or if necessary morning and evening. Metformin can also be used in combination with any other class of antidiabetic agent including insulin, and several fixed dose combination tablets are available in which metformin is combined with either a sulfonylurea or thiazolidinedione or dipeptidyl peptidase 4 (DPP-4) inhibitor (see below). Recall that although metformin alone is unlikely to cause serious hypoglycemia, this can occur when metformin is used in combination with an insulin-releasing agent or insulin.
During long-term use of metformin it is advisable to check at least annually for emergence of contraindications, particularly renal (as a minimum, serum creatinine). Metformin can reduce gastrointestinal absorption of vitamin B12, and although this is rarely a cause of frank anemia, an annual hemoglobin measurement is recommended especially for individuals with known or suspected nutritional deficiencies. Metformin should be temporarily stopped when using intravenous radiographic contrast media, or during surgery with general anesthesia or other intercurrent situations in which the exclusion criteria could be invoked. Substitution with insulin may be appropriate at such times.
Efficacy
As monotherapy in patients who are not adequately controlled by lifestyle management, optimally titrated metformin typically reduces fasting plasma glucose by 2–4 mmol/L, corresponding to a decrease in HbA1c by approximately 1–2% (11–22 mmol/mol) [39]. This is largely independent of body weight, age and duration of diabetes provided that some β-cell function is still present. To accommodate the progressive nature of T2DM, it is likely that uptitration of dosage and addition of a second agent will be required to maintain glycemic control in the long term.
Metformin carries minimal risk of significant hypoglycemia or weight gain when used as monotherapy. It may lead to a reduction of basal insulin concentrations, notably in hyperinsulinemic patients, which should help to improve insulin sensitivity. Minor improvements in the blood lipid profile have been observed during metformin therapy, mostly in hyperlipidemic patients: plasma concentrations of triglycerides, fatty acids and low density lipoprotein (LDL) cholesterol tend to fall, whereas high density lipoprotein (HDL) cholesterol tends to rise [37]. These effects appear to be independent of the antihyperglycemic effect, although a lowering of triglyceride and free fatty acids is likely to help improve insulin sensitivity and benefit the glucose–fatty acid cycle.
In the UKPDS, overweight patients who started oral antidiabetic therapy with metformin showed a 39% reduced risk of myocardial infarction compared with conventional treatment (P = 0.01) [40]. There was no obvious relationship with metformin dosage, suggesting that patients who can only tolerate a low dosage of metformin may benefit from continuing the drug, even when other agents are required to achieve adequate glycemic control. The decrease in myocardial infarction was not related to the extent of the glucose-lowering effect of metformin, or effects on classic cardiovascular risk factors such as blood pressure or plasma lipids. Reported benefits of metformin on various atherothrombotic risk markers and factors have been reported, including reduced carotid intima-media thickness (cIMT) (Table 29.6), increased fibrinolysis and reduced concentrations of the antithrombolytic factor plasminogen activator inhibitor-1 (PAI-1) [34]. Detracting somewhat from the generally favorable cardiovascular risk reports there is evidence that combination of metformin with a sulfonylurea may initially increase cardiovascular mortality [40,41]. One potentially confounding factor might be greater cardiovascular risk caused by more severe metabolic disease in patients needing treatment with the combination [42]. Evidence from large databases with sulfonylurea plus metformin combination therapy have been reassuring [43,44]. Indeed, recent results from the post-trial follow-up of UKPDS (without continuation of randomized groups) showed that the cardiovascular benefits of metformin were maintained [14].
When metformin is added to the regimens of patients receiving insulin therapy, a reduction of insulin dosage is often required, consistent with the ability of metformin to improve insulin sensitivity. Similarly, addition of insulin in patients already receiving metformin usually requires lesser dosages of insulin and results in less weight gain. Lesser amounts of insulin are also associated with fewer and less severe episodes of hypoglycemia [44,45]. Although metformin is not indicated for the prevention of diabetes, it is noteworthy that the US Diabetes Prevention Program found metformin to reduce the incidence of new cases of diabetes in overweight and obese subjects with impaired glucose tolerance by 33%, compared with a reduced risk of 58% using an intensive regimen of diet and exercise [46]. The preventive effect of metformin was most evident amongst younger, more obese individuals.
Adverse effects
The main tolerability issue with metformin is abdominal discomfort and other gastrointestinal adverse effects, including diarrhea. These are often transient and can be ameliorated by taking the drug with meals and titrating the dose slowly. Symptoms may remit if the dose is reduced, but around 10% of patients cannot tolerate the drug at any dose. The most serious adverse event associated with metformin is lactic acidosis; it is rare (probably about 0.03 cases per 1000 patient-years), but about half of cases are fatal [31,47,48]. Because the background incidence of lactic acidosis amongst patients with T2DM has not been established, it is possible that some cases previously attributed to metformin were caused by other factors.
Most reported cases of lactic acidosis in patients receiving metformin have been caused by inappropriate prescription, particularly overlooking renal insufficiency. The resulting accumulation of metformin is likely to increase lactate production, and increasing lactate will be aggravated by any hypoxic condition or impaired liver function. Hyperlactatemia occurs in cardiogenic shock and other illnesses that decrease tissue perfusion, so metformin may only be an incidental factor in some cases. Nevertheless, metformin should be stopped immediately in all cases of suspected or proven lactic acidosis, regardless of cause.
Lactic acidosis is typically characterized by a raised blood lactate concentration (e.g. >5 mmol/L), decreased arterial pH and/or bicarbonate concentration with an increased anion gap ([Na+] – [Cl– + HCO3–] >15 mmol/L). Presenting symptoms are generally non-specific, but often include hyperventilation, malaise and abdominal discomfort. Treatment should be commenced promptly without waiting to determine whether metformin is a cause; bicarbonate remains the usual therapy, but evidence of its efficacy is limited. Hemodialysis to remove excess metformin can be helpful, and may assist restoration of fluid and electrolyte balance during treatment with high-dose intravenous bicarbonate.
Sulfonylureas
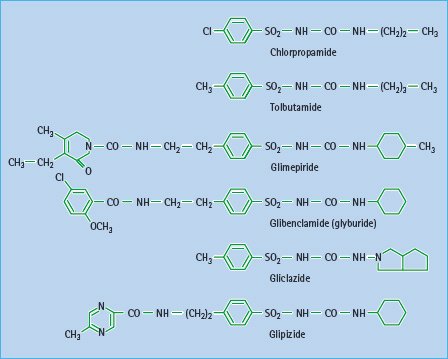
Since their introduction in the 1950s, sulfonylureas have been used extensively as insulin secretagogues for the treatment of T2DM. Sulfonylureas were developed as structural variants of sulfonamides after the latter were reported to cause hypoglycemia [49]. Early sulfonylureas such as carbutamide, tolbutamide, acetohexamide, tolazamide and chlorpropamide are often referred to as “first generation.” These have been largely superceded by more potent “second-generation” sulfonylureas, notably glibenclamide (glyburide), gliclazide, glipizide and most recently glimepiride (Figure 29.6).
Mode of action
Sulfonylureas act directly on the β-cells of the islets of Langerhans to stimulate insulin secretion (Figure 29.7). They enter the β-cell and bind to the cytosolic surface of the sulfonylurea receptor 1 (SUR1), which forms part of a transmembrane complex with ATP-sensitive Kir6.2 potassium channels (K+ATP channels) (Figure 29.8) [50,51]. Binding of a sulfonylurea closes the K+ATP channel, reducing the efflux of potassium and enabling membrane depolarization. Localized membrane depolarization opens adjacent voltage-dependent L-type calcium channels, increasing calcium influx and raising the cytosolic free calcium concentration. This activates calcium-dependent signaling proteins that control the contractility of micotubules and mictrofilaments that mediate the exocytotic release of insulin granules. Preformed insulin granules adjacent to the plasma membrane are promptly released (“first phase” insulin release), followed by a protracted (“second phase”) period of insulin release that begins about 10 minutes later [52]. The “second phase” of insulin release involves translocation of preformed and newly formed insulin granules to the plasma membrane for secretion. Sulfonylureas continue to stimulate insulin release while they are bound to the SUR1 provided that the β-cells are functionally competent. Some desensitization, however, occurs during repeated and protracted stimulation [53]. Because sulfonylureas can stimulate insulin release when glucose concentrations are below the normal threshold for glucose-stimulated insulin release (approximately 5 mmol/L), they are capable of causing hypoglycemia, mainly because of insulin-induced suppression of hepatic glucose production.
Figure 29.7 Sulfonylureas act on the pancreatic β-cell to stimulate insulin secretion. They bind to the cytosolic surface of the sulfonylurea receptor 1 (SUR1), causing closure of ATP-sensitive Kir6.2 potassium channels, depolarizing the plasma membrane, opening calcium channels, and activating calcium-dependent signaling proteins that control insulin exocytosis.
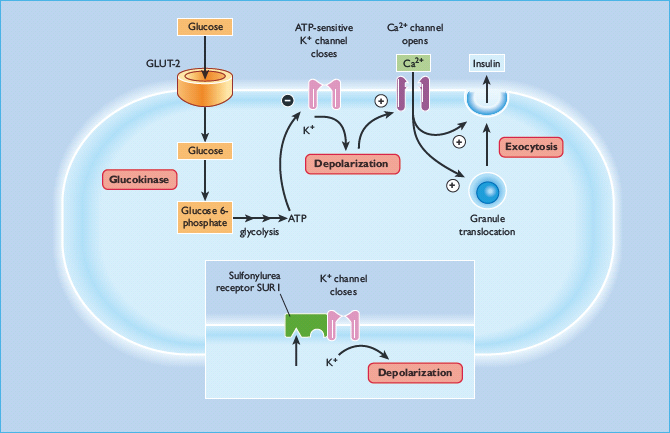
Figure 29.8 This illustrates the transmembrane complex of the SUR1 sulfonylurea receptor and the ATP-sensitive Kir6.2 potassium efflux channel on the pancreatic β-cells (a). Each SUR1 has a cytosolic sulfonylurea (S) binding site and a benzamido (B) binding site. SUR2A on cardiac muscle cells (and SUR2B on vascular smooth muscle cells) does not have a sulfonylurea binding site (b). The SUR1 –Kir6.2 complex is a non-covalently bonded octamer comprising 4 * SUR1 and 4 * Kir6.2, illustrated from the cytosolic surface to show the sulfonylurea and benzamido binding sites (c). The Kir6.2 molecules are located at the centre and form the K+ efflux pore. The Kir6.2 channel has a cytosolic binding region for ADP/ATP.
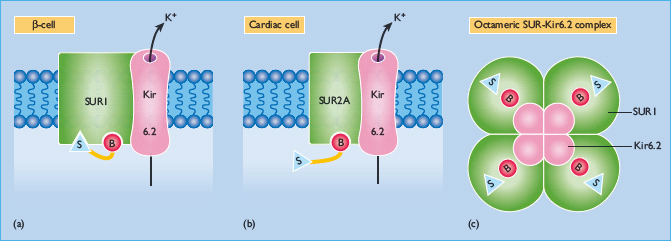
There is some evidence that sulfonylureas exert minor glucose-lowering effects independently of increased insulin secretion [54,55]. A small reduction in glucagon concentrations, increased peripheral glucose transport and increased hepatic glucose deposition have been reported, but these effects are not regarded to be of sufficient magnitude to be clinically relevant.
Pharmacokinetics
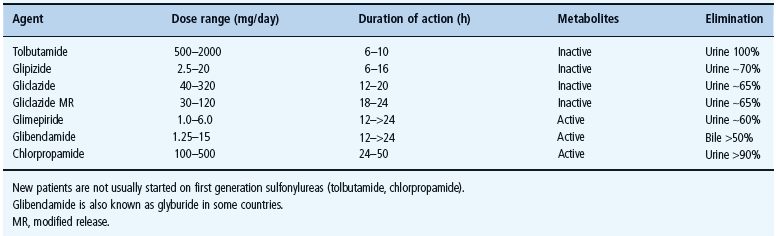
Sulfonylureas vary considerably in their pharmacokinetic properties (Table 29.7), which in turn affects their clinical suitability for different patients [54,55,56]. They are generally well absorbed, and reach peak plasma concentration in 2–4 hours. Sulfonylureas are highly bound to plasma proteins which can lead to interactions with other protein-bound drugs such as salicylates, sulfonamides and warfarin. Also, displacement of protein-bound sulfonylurea can increase the risk of hypoglycemia (Table 29.5). Sulfonylureas are metabolized in the liver to varying extents to a range of active and inactive metabolites that are eliminated along with unchanged drug via the bile and urine. Longer acting sulfonylureas can be given once daily but carry greater risk of hypoglycemia, especially with active metabolites. Sites and rates of metabolism and elimination are also important considerations, especially in older patients and individuals with coexistent liver or kidney disease or taking several other medications.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


