Summary of key facts
- •
Immune checkpoints are molecular pathways that blunt the antitumor immune response via limiting the creation of cytotoxic T cells and triggering exhaustion of tumor-reactive immune cells.
- •
Immune checkpoint inhibitors, especially those targeting the PD-1/PD-L1 pathway, are used to treat patients with a wide range of cancers.
- •
Adoptive transfer of TILs can result in clinical responses in patients with metastatic melanoma.
- •
T cells can be engineered to express a CAR targeting a range of cell surface antigens.
- •
CD19-specific CAR T cells can provide substantial benefit to patients with B-cell malignancies.
- •
Monoclonal antibodies can treat cancer by inducing antibody-dependent cellular cytotoxicity or complement-dependent cytotoxicity.
- •
Antibody–drug conjugates allow for antigen-specific delivery of a cancer therapeutic including cytotoxic agents, immunotoxins, and radiopharmaceuticals.
- •
The success of prophylactic cancer vaccines against human papilloma virus and hepatitis B have fostered interest in using vaccines to prevent other viral-associated malignancies.
- •
An autologous dendritic cell vaccine targeting prostatic acid phosphatase was the first therapeutic cancer vaccine approved for use in patients with advanced cancer.
- •
Building on the successes of cancer immunotherapy, trials of combinatorial immunotherapy are underway to assess whether treatment with multiple agents can further augment the antitumor immune response.
The role and application of checkpoint blockade
Introduction
Among the clinical successes of cancer immunotherapy, the strategy of immune checkpoint blockade has been adopted most broadly. Immune checkpoints refers to the molecular pathways responsible for blunting the antitumor immune response. These include mechanisms that limit the creation of cytotoxic T cells and that result in exhaustion of tumor-reactive immune cells. Though a range of approaches directed toward unique vulnerabilities of a specific cancer hold promise, the targeted manipulation of common molecular pathways that suppress the effector arm of the immune system has demonstrated efficacy across many malignancies. This benefit has been realized through clinical development of monoclonal antibodies that disinhibit the immune system, thereby fueling excitement for pharmacologic manipulation of additional immune checkpoints and for use of checkpoint blockade in combination with other cancer therapeutics.
Background
Attempts to generate an immune response against cancer have focused on creation of tumor-reactive T cells and sustained activation of these cytotoxic effector cells. Early efforts were aimed at identification of tumor-associated antigens with the goal of immunizing a host against these peptides. Related work sought to expand the number of cytotoxic T cells capable of recognizing and destroying cancer cells. Through this work, several mechanisms were identified as key mediators by which tumors evade the immune system. Among these, cytotoxic T-lymphocyte antigen 4 (CTLA-4, CD152), an inhibitory signal in the immune synapse, proved to be important. Blockade of the CTLA-4 receptor was shown to induce tumor regression in preclinical models. Furthermore, clinical use of anti-CTLA-4 therapy provided benefit to patients with advanced cancer, thereby leading to U.S. Food and Drug Administration (FDA) approval in 2011. Likewise, development of pharmacologic agents blocking additional immune checkpoints have been successful. Most notable, inhibition of the programmed death 1 (PD-1, CD279) receptor and its primary ligand programmed death ligand-1 (PD-L1, CD274, B7-H1) has shown even greater efficacy across a wide range of malignancies. The success of immune checkpoint blockade was recognized by awarding the 2018 Nobel Prize in Physiology or Medicine to James Allison for work on CTLA-4 and Tasuku Honjo for work on PD-1. Their discoveries have revolutionized the field of immunotherapy, leading to research exploring other immune checkpoint pathways and ways to combine checkpoint blockade with other cancer treatments.
Cytotoxic T-lymphocyte antigen 4
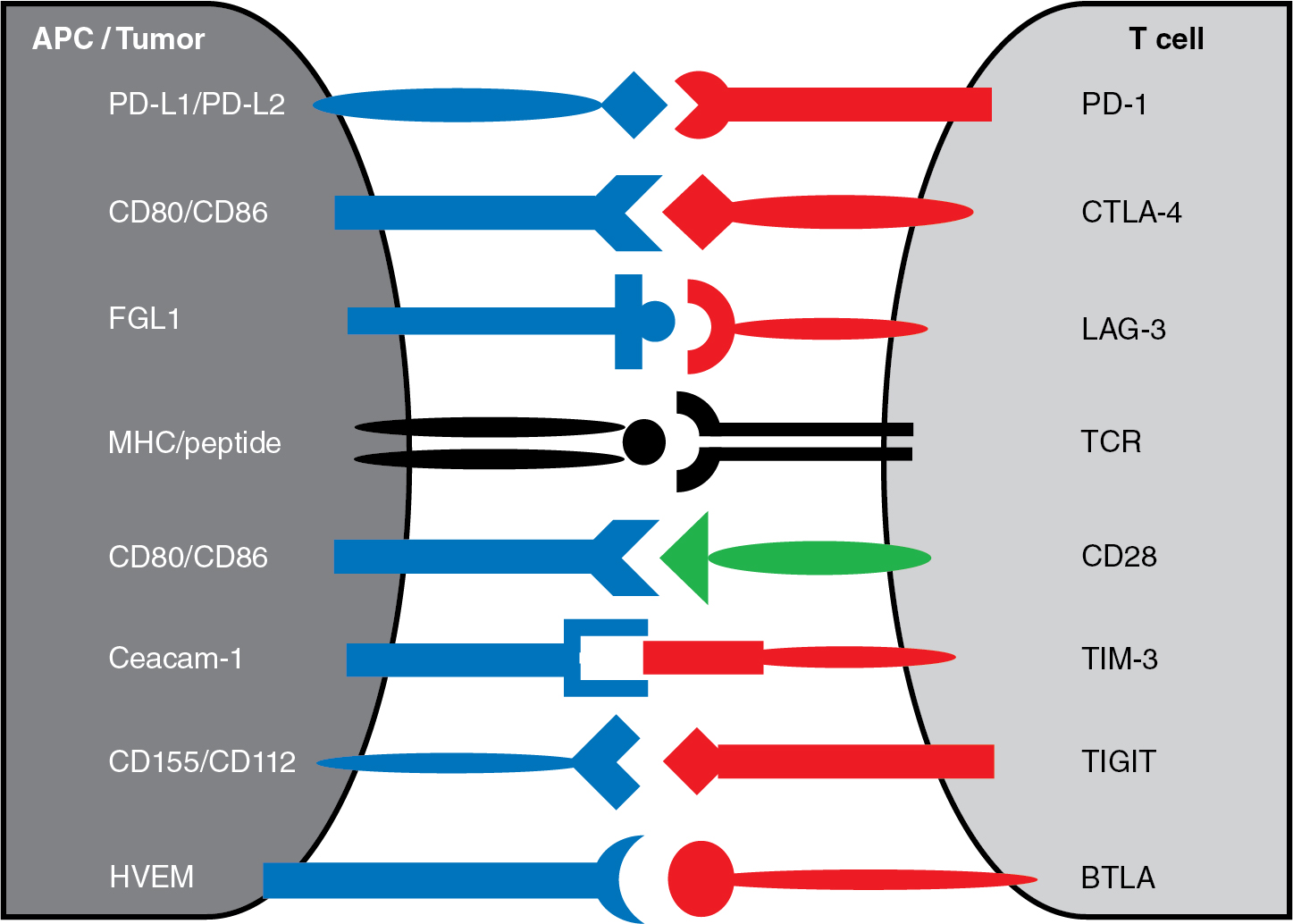
As outlined in Chapter 7 , tumor-reactive T cells are generated when naïve T cells receive two signals from antigen-presenting cells. There must be interaction between the T-cell receptor and its cognate peptide–major histocompatibility complex (MHC; signal 1) and engagement of the costimulatory receptor CD28 with one of its ligands CD80 or CD86 (signal 2), as depicted in Fig. 10.1 . Though early efforts in the development of cancer immunotherapies often focused on signal 1, interest in targeting signal 2 was bolstered by the clinical success of inhibiting the negative costimulatory receptor CTLA-4. Compared with CD28, CTLA-4 has a greater affinity for CD80 and CD86 (see Fig. 10.1 ) and can outcompete CD28 for these costimulatory ligands, thereby impairing T-cell activation. Furthermore, CTLA-4 has the potential to dampen antitumor immune responses, because CTLA-4 is absent on resting memory T cells but becomes rapidly expressed after T-cell activation. This feedback inhibition likely facilitates immune tolerance but also allows tumors to evade the immune system.
The seminal observation that CTLA-4 blockade can potentiate the antitumor immune response came from experiments in which administration of anti-CTLA-4 antibodies led to rejection of transplanted murine colon carcinoma in immunocompetent mice. This preclinical success was followed by clinical development of monoclonal antibodies targeting CTLA-4. Ipilimumab (Yervoy, Bristol-Myers Squibb) was approved by the FDA in 2011 for the treatment of advanced melanoma based on results of two phase III clinical trials. , Further studies have shown efficacy in subsets of patients with a range of cancers including surgically resected melanoma, renal cell carcinoma, colorectal cancer, hepatocellular carcinoma, and non-small cell lung cancer. Though the potential effect of targeting CTLA-4 was clear from these trials, the magnitude of clinical benefit derived from ipilimumab monotherapy was modest. Clinical data pooled from studies of ipilimumab-treated patients with advanced melanoma reported a median overall survival of approximately 1 year (11.4 months) and fewer than 20% of patients had long-term survival. Another CTLA-4-blocking monoclonal antibody tremelimumab (Pfizer, MedImmune, Astra Zeneca) was evaluated in patients with advanced melanoma and demonstrated a similar median overall survival of approximately 1 year (12.6 months); this was compared with standard-of-care chemotherapy, which had a median overall survival of 10.7 months, and the small improvement in survival was not statistically significant (hazard ratio = 0.88, P = .13). Interestingly, the duration of response was substantially longer in patients receiving tremelimumab compared with standard-of-care chemotherapy (35.8 vs 13.7 months, P = .001).
Taken in sum, data from these clinical trials exploring the activity of anti-CTLA-4 therapy led to two fundamental conclusions. First, blockade of an immune checkpoint like CTLA-4 has potential value to patients with a spectrum of human malignancies. Whereas many treatment regimens are only effective against one tumor type, targeting a molecular immune pathway allows broader utility across a range of cancers. Second, despite low response rates with anti-CTLA monotherapy, patients who benefit from this treatment often have favorable long-term outcomes not seen with conventional chemotherapy. This difference seen at the tail of the survival curve is exciting to patients, clinicians, and scientists alike. Therefore it is not surprising that the early experience with CTLA-4 blockade fueled efforts to target other immune checkpoint pathways, to identify predictive biomarkers of response, and to combine CTLA-4 blockade with other cancer therapies.
Programmed death 1
Subsequent to the promising results from clinical trials using anti-CTLA-4 antibodies, there was rapid development of agents targeting the PD-1 pathway. The PD-1 receptor is similar to CTLA-4 in that both are expressed on activated T cells and can transduce inhibitory signals when engaged with their ligands. However, these immune checkpoint pathways have important differences. For example, the known ligands for PD-1 are PD-L1 (CD274, B7-H1) and PD-L2 (CD273, B7-DC) (see Fig. 10.1 ). Unlike the CTLA-4 ligands that are expressed on antigen-presenting cells, the PD-1 ligands are found on tumors and other cells within the tumor microenvironment. It is believed that PD-L1 is the dominant ligand present on solid tumors, whereas PD-L2 is found at higher levels on hematologic malignancies. Either ligand can inhibit activated T cells that express PD-1. Whereas the CTLA-4 pathway is generally felt to mediate feedback inhibition between antigen presenting cells and effector T cells in secondary lymphoid tissues, the PD-1 pathway is believed to block tumor-reactive T cells within the tumor microenvironment. Regardless of the precise mechanism(s), PD-1 seems to promote T-cell exhaustion and dampen the antitumor immune response.
Much of our insight into the PD-1 pathway stems from preclinical testing. Notably, PD-L1 expression on tumor cells was shown to confer resistance to host immunity. Furthermore, blockade of the PD-1 ligand inhibited tumor growth in this mouse model of myeloma. Likewise, administration of a monoclonal antibody to PD-L1 in combination with adoptive transfer of tumor-reactive T cells resulted in long-term survival of squamous cell carcinoma-bearing mice. Beyond its promise of clinical benefit, mouse models also suggested that targeting the PD-1 pathway may have less toxicity than targeting other immune checkpoints. Whereas CTLA-4 knockout mice develop uncontrolled lymphoproliferation and have early lethality, PD-1 knockout mice have milder and delayed-onset autoimmunity. Based on these encouraging findings, therapies targeting the PD-1 pathway were advanced into human trials.
Early clinical success was seen with the anti-PD-1 monoclonal antibody nivolumab (Opdivo, Bristol-Myers Squibb). In a Phase I dose escalation study of 39 patients, there were objective responses seen in 3 patients with melanoma, renal cell carcinoma, and colorectal cancer. Subsequent studies with nivolumab have shown durable benefit in patients with non-small cell lung cancer, renal cell carcinoma, and melanoma, as well as efficacy in a number of other cancers; therefore nivolumab received FDA approval in 2014. Of note, pembrolizumab (Keytruda, Merck) was the first monoclonal antibody targeting PD-1 approved by the FDA. This followed impressive results from a large early-phase study in which clinical responses were seen in 52 of 135 patients with advanced melanoma who received the anti-PD-1 antibody (originally named lambrolizumab), including patients previously treated with the anti-CTLA-4 monoclonal antibody ipilimumab.
The efficacy of pembrolizumab was not limited to melanoma, because its use has benefit in the treatment of groups of patients with bladder cancer, breast cancer, cervical cancer, colorectal cancer, gastroesophageal cancers, head and neck squamous cell carcinoma, hepatocellular carcinoma, lymphoma, Merkel cell carcinoma, non-small cell lung cancer, renal cell carcinoma, and tumors with high mutational burden or deficiencies in mismatch repair. Building on the success of nivolumab and pembrolizumab, several other agents targeting the PD-1/PD-L1 pathway have been approved. This includes cemiplimab (Libtayo, Sanofi/Regeneron), which has shown efficacy in the treatment of advanced nonmelanoma skin cancers and PD-L1-expressing non-small cell lung cancer. There has also been clinical success with anti-PD-L1 therapies, including FDA approval of avelumab (Bavencio, Pfizer), atezolizumab (Tecentriq, Genentech), and durvalumab (Imfinzi, Astra Zeneca). With many more trials underway and additional agents in clinical development, blockade of the PD-1 immune checkpoint pathway continues to be a key target in cancer therapy.
Lymphocyte activation gene 3
Subsequent to the clinical success of PD-1 and CTLA-4 pathway blockade, the LAG-3 (CD223) immune checkpoint has become another target of interest in cancer immunotherapy. LAG-3 is a type I transmembrane protein and member of the immunoglobulin superfamily, which is expressed on lymphocytes and binds MHC class II. Early interest in LAG-3 stemmed from its ability to enhance the function of antigen-presenting cells and thereby create a more robust antitumor immune response. Tumor cells engineered to express LAG-3 had impaired growth compared with native tumor cells when implanted into immunocompetent mice, and these mice were protected from future challenge with native tumor cells. This strategy of enhancing host immunity against cancer led to the development of soluble LAG-3 as a vaccine adjuvant. The first agent tested in clinical trials was eftilagimod alpha (IMP321, Immutep), a recombinant protein combining the LAG extracellular domains with the Fc domain of human immunoglobulin (Ig) G1. Patients with metastatic renal cell carcinoma were treated with escalating doses of the compound, which led to activation of CD8 T cells and reduced tumor growth in some patients. Subsequent clinical trials combined eftilagimod alpha and cytotoxic chemotherapy, suggesting modest clinical benefit in patients with metastatic breast cancer. Studies evaluating soluble LAG-3 with cancer vaccines or in combination with PD-1 blockade are ongoing.
In addition to its ability to bind MHC class II, LAG-3 can interact with fibrinogen-like protein 1 (FGL1) (see Fig. 10.1 ). This distinct function is believed to trigger T-cell exhaustion and blunt T-cell proliferation within the tumor microenvironment, mimicking the interaction between PD-1 and its ligands. Interestingly, LAG-3 and PD-1 are both overexpressed by immune cells in models of chronic infection and LAG-3 expression often parallels PD-1 expression in tumor-reactive T cells. Furthermore, mice deficient in both LAG-3 and PD-1 develop extensive lymphocytic infiltration that results in early death. Based on the clinical success of anti-PD-1 therapy and the potential synergy between the two pathways, preclinical evaluation of LAG-3 blockade has focused on combinatorial approaches with anti-PD-1 agents. Encouragingly, combined anti-PD-1 and anti-LAG-3 treatment has been effective in mouse cancer models. The anti-LAG-3 monoclonal antibody relatlimab (BMS-986016, Bristol-Myers Squibb), which was approved by the FDA in 2022, has been tested in a large phase I/II trial with or without nivolumab in the treatment of patients with advanced cancer. There were encouraging results from this study (NCT01968109) in patients with melanoma, including those previously treated with anti-PD-1/anti-PD-L1 therapy. A subsequent randomized phase II/II trial of nivolumab with or without relatlimab demonstrated greater progression-free survival from dual checkpoint inhibition in patients with previously untreated melanoma. With additional studies ongoing and novel agents in development, it seems that therapies targeting LAG-3 will have a promising role in the treatment of cancer.
Other immune checkpoint pathways
As the understanding of other immune checkpoints pathways grows, there is interest in clinical development of new agents to expand the armamentarium of cancer therapy. Though a comprehensive summary is beyond the scope of this work, a general overview of additional promising targets is provided in the following sections.
T-cell immunoglobulin and mucin domain–containing 3
TIM-3 (CD366) is an immunoglobulin superfamily receptor expressed on the surface of immune cells. Like other immune checkpoint molecules discussed previously, research has focused on its function in T cells. TIM-3 has several known ligands, each seemingly capable of distinct cellular effects. For example, galectin-9 is a soluble ligand that has been shown to induce apoptosis of TIM3 + T cells and another soluble ligand high-mobility group protein B1 (HMGB1) can interact with TIM-3 + dendritic cells to dampen antitumor immunity. Nonetheless, it is the interaction between TIM-3 and membrane-bound carcinoembryonic antigen cell adhesion molecule 1 (Ceacam-1) that is believed to trigger T-cell inhibition (see Fig. 10.1 ). Preclinical models have shown that tumor-infiltrating lymphocytes expressing TIM-3 and PD-1 have reduced ability to proliferate and secrete proinflammatory cytokines; furthermore, dual blockade of TIM-3 and PD-1 had therapeutic benefit in mouse tumor models. Therefore clinical development of monoclonal antibodies blocking TIM-3 has largely focused on combination with anti-PD-1 therapy with some trials even exploring coadministration of agents blocking PD-1, LAG-3, and TIM-3. In addition, bispecific antibodies simultaneously targeting PD-1 and TIM-3 are under investigation.
T-cell immunoreceptor and ITIM domain
Sharing structural domains with other immune checkpoints molecules discussed previously, TIGIT was identified through a genomic search for cell surface proteins containing a conserved immunoreceptor tyrosine-based inhibitory motif (ITIM) domain. TIGIT is expressed on T cells and can trigger the immunosuppressive function of dendritic cells through engagement with its ligand CD155. Further research revealed that TIGIT also serves as an immune checkpoint with the ability to suppress antitumor T-cell function. This can occur through two related mechanisms as (1) TIGIT provides inhibitory signaling upon binding its ligands and (2) TIGIT impairs excitatory signaling from CD226 by outcompeting this costimulatory receptor for the shared ligands CD155 and CD112 (see Fig. 10.1 ); the relationship between TIGIT and CD226 is akin to that of CTLA-4 and CD28. Likewise, TIGIT blockade can enhance antitumor responses in animal models, and this effect is augmented by combination with other immune checkpoint inhibitors. Based on these encouraging results, clinical evaluation of anti-TIGIT agents is currently underway with several studies targeting TIGIT in addition to other checkpoint pathways.
B- and T-lymphocyte attenuator
B- and T-lymphocyte attenuator (BTLA, CD272) is an inhibitory receptor in the CD28 superfamily. As the name suggests, it is expressed on B cells and T cells. However, BTLA can also be found on macrophages and dendritic cells. Herpes virus entry mediator (HVEM) is recognized as the primary ligand for BTLA (see Fig. 10.1 ); HVEM expression can lead to inhibition of T-cell proliferation and function. Like other immune checkpoint molecules, BTLA has been identified on tumor-infiltrating lymphocytes across a range of malignancies. Given its potential role in dampening the antitumor immune response, clinical development of agents targeting BTLA are proceeding, with phase I clinical trials for patients with advanced cancer now underway.
Conclusions
Immune checkpoint blockade has rapidly become one of the most exciting cancer treatments. Building on the pioneering work of Nobel laureates James Allison and Tasuku Honjo, scientists continue to develop new agents that can augment the antitumor immune response. Whether through promoting generation of tumor-reactive immune cells or preventing exhaustion of cytotoxic T cells, targeting these molecular pathways has the potential to treat a broad range of malignancies. The clinical success of anti-CTLA-4 therapy provided a glimpse of this promise. However, the broad efficacy of targeting the PD-1 pathway has been transformational. As such, clinicians continue to evaluate these new therapies and explore rational combinations to maximize the clinical benefit for patients with cancer.
The role and application of cell-based therapies
Case vignette
A 17-year-old male presents in the clinic with subjective fever and easy bruising for the past week. After appropriate workup a diagnosis of t(9;22)/BCR-ABL1 negative pre-B cell acute lymphoblastic leukemia (ALL) is made. Induction therapy with vincristine, anthracycline, asparaginase, and steroids fails to result in complete remission. Additional options for achieving remission to enable hematopoietic cell transplantation (HCT) are discussed with the patient and family, and treatment with tisagenlecleucel, a chimeric antigen receptor (CAR) T-cell product, is scheduled. The patient’s lymphocytes are collected, the autologous CAR T cell product is prepared, and infusion is performed after administration of preparative lymphodepleting chemotherapy. On the fourth day postinfusion the patient develops fever (38.9°C), hypotension (BP 73/35), and hypoxia (Sa o 2 87%), concerning for cytokine release syndrome (CRS). Intravenous fluids, oxygen, vasopressors, and tocilizumab (interleukin [IL]-6 antagonist) are given, with rapid improvement in the patient’s vital signs. Subsequent disease monitoring of the bone marrow and spinal fluid is significant for a lack of detectable disease. This case demonstrates the induction of remission for a poor prognosis malignancy after adoptive transfer of CAR T cells, a revolution in the field of immunotherapy.
Tumor-infiltrating lymphocytes
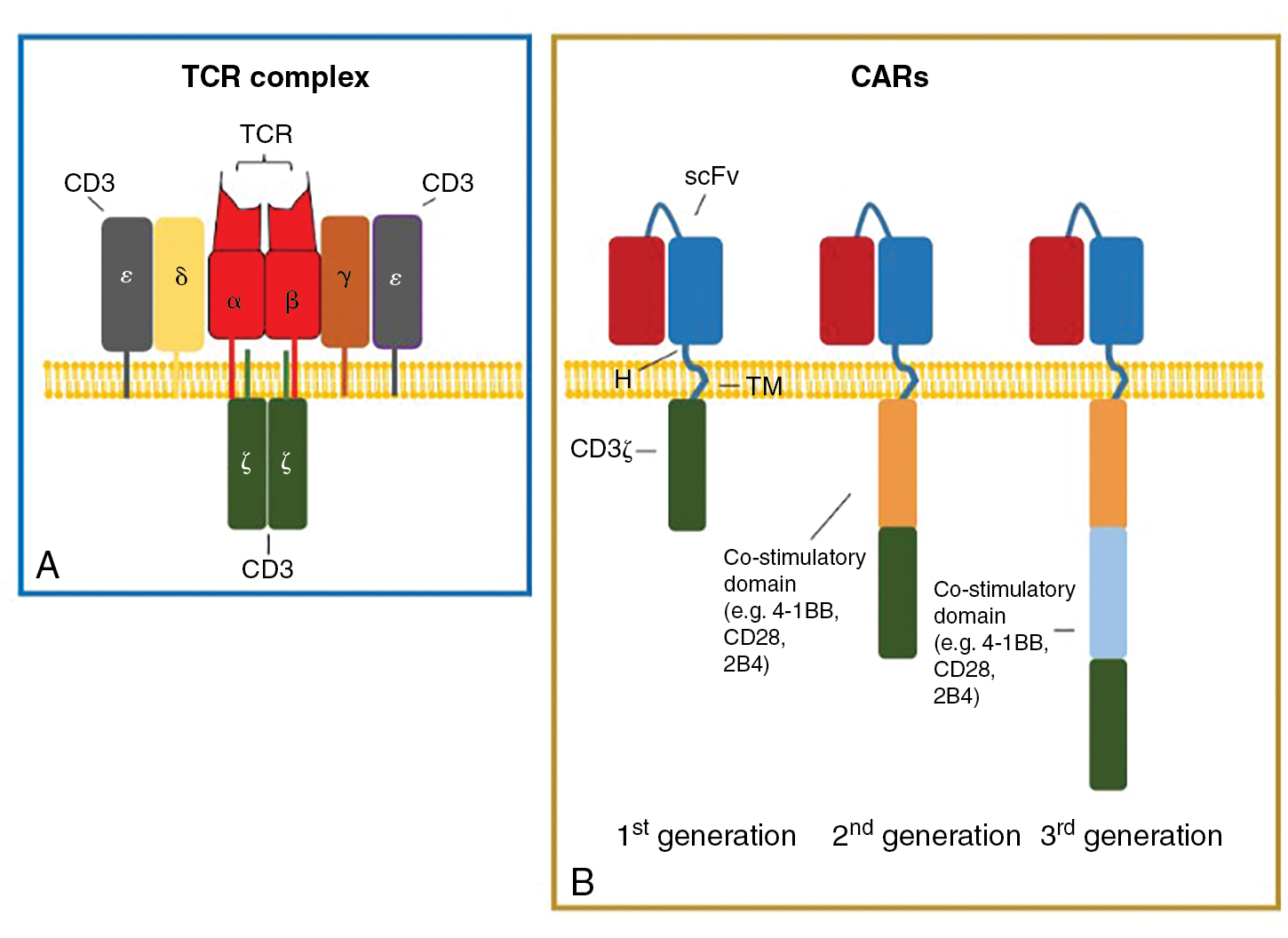
The immune system is populated by important effector and supportive cells that react to stressed, infected, or transformed cell types. T lymphocytes are likely the most important, because they quickly traffic to and invade tumor sites and then are able to elicit a specific immune response after T-cell receptor (TCR) recognition of tumor-specific antigens presented in the context of MHC ( Fig. 10.2 ). These infiltrating cells are commonly referred to as tumor-infiltrating lymphocytes (TILs). When functional TILs can be identified and directly isolated from a patient’s tumor, this is generally considered a favorable prognostic factor. In addition, isolated TILs may be purified and expanded for use in adoptive cell transfer (ACT) as treatment against a variety of solid tumors. Marrow-infiltrating lymphocytes can also be harvested and used against liquid cancers, like multiple myeloma, in which the bone marrow is considered the tumor microenvironment. Rosenberg et al. were the first to investigate the role of TILs as adoptive immunotherapy for cancer in 1986. TILs isolated from tumor-engrafted C57BL/6 mice and cultured in IL-2 for 15 days were 50 to 100 times more potent than lymphokine-activated killer (LAK) cells (widely used in preclinical and clinical studies in 1980s) against various tumor micrometastases in animal models. A year later, Kradin et al. performed the first clinical trial of TILs, showing safety in administration but no responses in seven patients with lung cancer. The first objective clinical responses in TIL treatment were seen in a cohort of 20 patients with metastatic melanoma by Rosenberg et al. in 1989. A single dose of cyclophosphamide followed by infusion of TIL and IL2 led to a reduction of melanoma lesions in 58% of patients, with complete response (CR) in 1 patient and partial response (PR) in 10 patients. This dramatic outcome led to many future clinical trials for metastatic melanoma and other cancers.
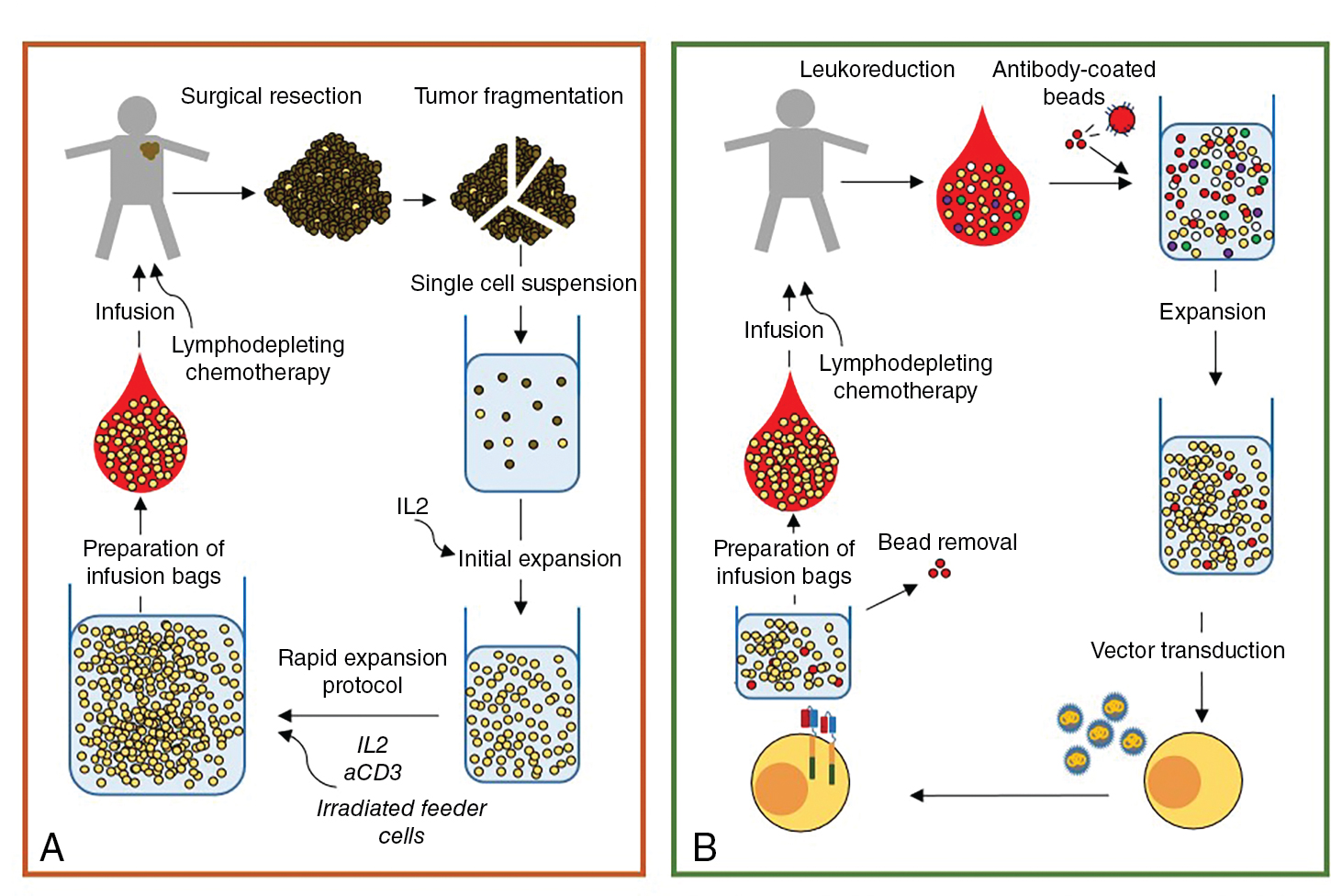
Generation of TIL is a multistep process ( Fig. 10.3 ). First, the tumor lesion is surgically excised and a single-cell suspension is prepared by mechanical or enzymatic separation. This suspension is cultured with IL-2 for approximately 2 weeks, leading to survival and proliferation of TILs but death of tumor cells. Next, TILs are rapidly expanded using IL-2, anti-CD3 antibody, and irradiated feeder cells. Once the necessary yield for immunotherapeutic infusion is achieved (commonly 1 × 10 10 to 2 × 10 11 cells), the TIL product consists of a heterogeneous population of T cells. Activated lymphocytes can be purified via flow cytometric or magnetic bead sorting if specific T-cell subsets are desired. Additional studies are needed to define the clinical benefit of TIL therapy and to identify the optimal T-cell population for sustained antitumor activity.
Since 1989, TIL therapy has been investigated in more than 15 clinical trials of patients with metastatic melanoma. Most of these studies have evaluated TILs as monotherapy in patients not previously treated with PD-1 blockade. Overall, an objective clinical response and a complete remission have been documented in approximately 50% and 10% to 15% of treated patients, respectively. As immune checkpoint inhibition has evolved to become first-line treatment, these encouraging results led several groups to investigate the potentially additional benefit of TILs as a combinational therapy with or as an alternative treatment. Sarnaik et al. investigated the role of the Lifileucel TIL product in the treatment of 66 patients with advanced melanoma (metastatic or unresectable) that progressed after immune checkpoint inhibition (anti-PD-1, anti-PD-L1, anti-CTLA-4) and targeted therapies (BRAF ± MEK inhibitors if BRAF V600 mutated tumors). An objective response was seen in 36% of patients (22 PR and 2 CR), and 44% had stable disease. Objective response was observed to be 41% in the cohort of patients with cancer refractory to anti-PD-1 or anti-PD-L1 therapies. Rates of survival greater than 1 year were 92% in patients who had an objective response to treatment and 38% in patients with stable disease. These encouraging results hold promise for the use of TILs in patients with treatment-refractory melanoma in whom therapeutic options are limited. Nonetheless, there are no TIL therapies approved by the FDA for melanoma.
TILs have been isolated and expanded from solid tumors other than melanoma; these include breast cancer, renal cell carcinoma, and gastrointestinal malignancies. In contrast to TILs isolated from melanoma, these products have shown variable antitumor efficacy in preclinical models. There are limited clinical studies of TIL monotherapy as treatment for nonmelanoma solid tumors and only one trial reporting objective responses. Stevanović et al. investigated the infusion of TILs selected for human papillomavirus (HPV) oncoprotein reactivity in nine patients with refractory, metastatic cervical cancer. Two patients achieved a CR (lasting 15 and 22 months) and one patient had a short-lived PR. The disparity in antitumor responsiveness of melanoma-derived TILs versus those obtained from other tumors may be explained by differences in (1) disease-associated mutational burden that ultimately drives immunogenicity, (2) the variable immunosuppressive tumor microenvironment, (3) differences in cellular composition within the harvested TILs, and (4) the differentiation state of infiltrative CD4 + and CD8 + T lymphocytes. Melanoma is characterized by high tumor mutational burden (TMB) and neoantigen expression, which correlates with clinical benefit of TIL immunotherapy. Furthermore, melanoma TILs are less differentiated and possess high proliferative capacity. Continued research efforts are imperative to overcome the limitations of TIL therapy and improve clinical responses in tumors other than melanoma.
Preconditioning with lymphodepleting (LD) chemotherapy prior to TIL infusion is crucial for optimal activity. Induction of lymphopenia creates “physical space” for the transferred TILs, depletes immunosuppressive regulatory T cells, and reduces competition for homeostatic cytokines (e.g., IL-7 and IL-15). One standard nonmyeloablative LD chemotherapy regimen includes cyclophosphamide (Cy, 60 mg/kg for 2 days, 120 mg/kg) and fludarabine (Flu; 25 mg/m 2 for 5 days, 125 mg/m 2 ) administered 24 hours before cell infusion. Nissani et al. investigated the efficacy and safety of different LD regimens including Cy, Flu, and/or total body irradiation (TBI) in patients with stage IV melanoma receiving TILs (NCT03166397). In this study, 60 mg/kg Cy/125 mg/m 2 Flu was the most optimal regimen to stimulate TIL expansion with a tolerable toxicity profile. LD is followed by TIL infusion and then commonly by systemic cytokine administration to support infused T-cell proliferation and survival. High-dose IL-2 is most frequently infused as a bolus of 720,000 IU/kg three times a day for 5 days, as tolerated. Lower doses of cytokine have been tested in small numbers of patients, but the potential to sustain satisfactory TIL responses requires investigation in larger cohorts of patients. Common toxicities observed with TIL treatment include febrile neutropenia, pancytopenia, high fever, chills, pulmonary congestion, and hypotension. These are likely associated with LD chemotherapy and/or IL-2 infusion. An uncommon, but significant, potential side effect of TIL therapy is the development of autoimmune-like side effects (e.g., vitiligo, uveitis, and rash).
The major advantage of TIL therapy over other forms of ACT is the capability of specific recognition and targeting of patient’s unique tumor-associated antigens. Exomic or whole-genome DNA sequencing with subsequent RNA sequencing have enabled identification of thousands of tumor-specific mutations and expression of neoantigens. Enrichment of the TIL product with neoantigen-directed populations can further enhance antitumor efficacy. This approach is possible by culturing TIL with autologous antigen-presenting cells that have been transduced with minigenes or peptides encoding these neoantigens. Despite its therapeutic promise, there are several challenges to widespread utilization of this technology. First, development of TIL therapy is personalized to a specific patient with high production costs and the need for specialized personnel and production facilities. Second, patients must undergo a procedure to obtain the tumor for TIL production. Third, long manufacturing time may limit its use in patients who experience tumor growth prior to administration of TIL therapy. For these reasons, the current focus of clinical development is centered on decreasing production time, improving patient selection, and simplifying manufacturing methods. Overcoming these limitations will help realize the promise of TIL therapy in the treatment of cancer.
Chimeric antigen receptor–engineered T cells
Adoptive transfer of T cells engineered to express a chimeric antigen receptor (CAR) has revolutionized the field of immunotherapy. Treatment with CAR T cells, which possess artificial receptors that enable robust, antigen-dependent T-cell activation, has resulted in clinical success of high-risk leukemia. CARs are synthetic molecules comprising an extracellular target-binding single-chain variable fragment (scFv) fused to a hinge, transmembrane, and intracellular domains that transmit downstream signaling (see Fig. 10.2 B). Eshhar et al. first engineered T cells with a chimeric molecule comprising a, scFV fused with the TCRζ chain. This molecule would later be described as a first-generation CAR (see Fig. 10.2 B). Despite antitumor specificity, T cells engineered with first-generation CARs did not expand in vivo or result in objective clinical responses after CAR-T cell infusion. Krause et al. first designed and tested a costimulatory-only CAR, using the CD28 intracellular domain. These CAR T cells effectively proliferated, produced IL-2, and exhibited antitumor killing in an antigen-specific manner. Savoldo et al. showed the positive clinical effect of including CD28 costimulation with TCRζ activation (signal 1 and signal 2) in CAR T cells infused to human patients. Use of costimulatory 4-1BB has also proven successful in amplifying CAR T-cell activation and survival. To date, CD28 and 4-1BB are the most well-studied costimulatory domains of T-cell CARs. Others, such as OX40 and ICOS, are also being tested.
Among all antigens targeted by CAR T cells, CD19 has thus far been most effectively targeted. CD19 is a molecule present on the surface of B-lineage malignant cells and is an optimal target because of its restricted expression in B cells (and no other healthy tissue) and lack of expression on hematopoietic stem cells and plasma cells. Kochenderfer et al. described the first report of a patient with relapsed stage IVB follicular lymphoma treated with a CD19.CD28.ζ CAR T-cells, showing lymphoma regression lasting 32 weeks. This report was followed by several clinical trials investigating the use of CD19 CAR T cells as treatment for relapsed or refractory pre-B cell ALL and non-Hodgkin lymphoma. Immunotherapy for relapsed or refractory pre-B cell ALL with CD19 CAR T-cells manufactured at a number of different academic centers led to complete disease remissions in 67% to 90% of patients. In non-Hodgkin lymphoma, Schuster et al. first showed impressive clinical outcomes in 28 patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma, with 64% achieving objective responses. This clinical success has quickly led to the development of commercially available CD19 CAR T cells for patients with CD19-positive malignancies.
In 2017, the FDA approved tisagenlecleucel, a CAR T-cell therapy that uses an anti-CD19.4-1BB.CD3ζ CAR (19.4-1BB.ζ), for the treatment of pediatric/young adult (≤25 years old) relapsed or refractory pre-B cell ALL. Tisagenlecleucel is the first cell-based gene therapy to gain commercial approval from the FDA, and its application was further expanded to include adult patients with relapsed or refractory DLBCL in 2018. To date, additional autologous CAR T-cell products have received FDA approval. Brexucabtagene autoleucel (19.CD28.ζ) is approved for use in adult patients with relapsed or refractory mantle cell lymphoma and ALL. Axicabtagene ciloleucel (19.CD28.ζ) and lisocabtagene maraleucel (19.4-1BB.ζ) are approved for adult relapsed or refractory B-cell lymphomas including DLBCL (not otherwise specified or arising from follicular lymphoma), primary mediastinal large B-cell lymphoma, and high-grade B-cell lymphoma. Lisocabtagene maraleucel is also used in adult relapsed or refractory grade 3B follicular lymphoma. Idecabtagene vicleucel is the first FDA-approved CAR T-cell product directed against a non-CD19 target: B-cell maturation antigen (BCMA). This BCMA.4-1BB.ζ CAR T-cell therapy is approved for the use in adult relapsed or refractory multiple myeloma, after treatment with four or more lines of therapies, to include an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
All of the FDA-approved CAR T-cell products use autologous patient-derived starting material. The use of autologous products is currently necessary to avoid the risk of graft-vs-host disease (GvHD), a potential severe side effect of allogeneic T-cell ACT. For CAR T-cell manufacturing, patient lymphocytes are collected and then enriched and activated, typically with anti-CD3/anti-CD28 antibody coated beads. T-cell activation is followed by cell modification, most often by viral transduction with vectors encoding the CAR transgene. After transduction, the cells are further expanded ex vivo to the desired cell number (i.e., 1–3 × 10 6 CAR T cells/kg), washed, and frozen for shipment and patient infusion (see Fig. 10.3 B). Similar to adoptive transfer of TILs, lymphodepleting chemotherapy is a pivotal component of CAR T-cell therapy, because this promotes in vivo expansion, long-term persistence, and antitumor effects. Lymphodepleting chemotherapy may also prevent CAR T-cell rejection due to immune responses against CAR epitopes. After completion of chemotherapy, patients are given intravenous CAR T-cell infusion and monitored daily for clinical signs of toxicity during the first 7 to 10 days.
CAR T-cell therapy is often associated with severe systemic toxicities that require a high level of clinical preparedness for prompt recognition and management. The most prevalent and often catastrophic side effect is cytokine release syndrome (CRS). CRS is characterized by systemic elevation of inflammatory cytokines resultant from CAR T-cell expansion and monocyte activation, including tumor necrosis factor alpha (TNF-α), IL-6, IL-1β, IL-10, and interferon gamma (IFN-γ). Clinical findings of CRS include high fever, flu-like symptoms, gastrointestinal (GI) disturbances, hypotension, tachycardia, hypoxia, capillary leak, organ failure, and death. Management is per CRS grade, defined by temperature elevation, severity of hypotension, and degree of hypoxia. With supportive measures, high-grade CRS (≥3) can usually be reversed with tocilizumab, a humanized monoclonal antibody against the IL-6 receptor. The use of high-dose corticosteroids is typically reserved for nonresponders to tocilizumab. Another serious complication of CAR T-cell therapy is immune effector cell–associated neurotoxicity syndrome (ICANS), most likely caused by blood–brain barrier (BBB) dysfunction in concert with T-cell activation. ICANS symptoms include headache, altered consciousness and delirium, expressive aphasia, seizure, and coma. Corticosteroids are the cornerstone of ICANS management, because blockade of the IL-6 receptor using tocilizumab is not effective because of limited pharmacologic distribution. Patients infused with CAR T cells at a higher disease burden are more likely to develop CRS or ICANS. Early intervention is important for resolution of these toxicities. However, these interventions to mitigate CRS or ICANS can also abrogate CAR T-cell function. A hemophagocytic lymphohistiocytosis-like toxicity has also been reported after CAR T-cell treatment associated with prominent hyperferritinemia and multiorgan failure. Other, less severe potential side effects of CAR T-cell treatment include hypogammaglobinemia due to B-cell aplasia (CD19 CARs), prolonged cytopenias, hypersensitivity reactions, serious infections, and, less commonly, second malignancies.
There are several areas in which clinical application of CAR T-cell immunotherapy that can be improved. The median time required for autologous CAR T-cell production is 23 days, a time frame that could be reduced through optimization of the manufacturing process. Moreover, per patient manufacturing costs are quite high, which is an important consideration that should stimulate efforts to automate and decrease cost so that all patients have access to this novel technology when it is needed. Continued research is also focused on strategies to mitigate observed toxicities, such as inclusion of safety switches that can be deployed when toxicity is anticipated. Improved CRS-directed therapies, such as with cytokine-directed monoclonal antibodies, is also an area of active clinical investigation. CAR T-cell application will ideally extend to solid tumors and other hematologic cancers (T-ALL, acute myeloid leukemia). Identification of novel tumor-specific antigens and modulation of interactions between T cells and the tumor microenvironment are specific areas under investigation in preclinical and clinical trials. With these advances, CAR T-cell technology is poised to expand the therapeutic options for patients with a range of cancer types.
The role and application of antibody-based therapies
Case vignette
A 13-year-old male presents to the emergency department with sore throat and fatigue. His CBC shows blasts on the differential and a comprehensive workup leads to the diagnosis of p53 mutated, complex karyotype acute myeloid leukemia (AML). He receives induction therapy with cytarabine, daunorubicin, and gemtuzumab ozogamicin, a CD33-targeting antibody–drug conjugate. His disease remains refractory to upfront treatment and salvage options are discussed with the family. He enrolls in a phase I clinical trial investigating the use of flotetuzumab, a CD123xCD3 dual-affinity retargeting protein (DART), for the treatment of pediatric recurrent or refractory AML. His treatment course is complicated by cytokine release syndrome requiring intensive care unit (ICU) admission for cardiorespiratory support. Disease assessment after one cycle of treatment demonstrated partial response with reduction in percentage of marrow blasts and negative CNS status. This case demonstrates the role of antibody-based therapies in the treatment of cancer, including monoclonal antibodies, antibody–drug conjugates, and novel application of bi- or trispecific engagers.
Background
Antibodies are a key component of the adaptive immune system. They are produced by mature B cells in a highly regulated way to diversify the immune response. B cells develop from lymphoid precursors in the bone marrow and through a series of maturation steps assemble unique antibodies on their cell surface. Antibodies are composed of two heavy chains and two light chains connected through disulfide bonds. The constant domains of the heavy chains form the Fc portion of the antibody. The variable regions of the heavy and light chain undergo a genetic process known as VDJ recombination to confer unique antigen specificity to each antibody. Mature B cells traffic to lymph nodes where, under antigen stimulation, they are selected to undergo class switching and further differentiate into plasma cells that release soluble antibodies into circulation. An antibody response is activated when the immune system recognizes a foreign antigen. This remarkable antigen specificity and diversity can be harnessed as immunotherapy to target tumor-specific surface antigens.
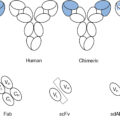
Monoclonal antibodies (mAbs) targeting tumor-specific antigens can be isolated for further design and translation for clinical use. Two primary platforms exist for the generation of mAbs with high antigen-specific recognition. One technology comprises injection of a tumor antigen into a nonhuman species and isolation of antibodies specific to the foreign antigen. These antibodies can then be engineered to create chimeric or humanized antibodies to minimize immunogenicity by incorporation of the nonhuman variable regions into a human Fc domain. Fully human antibodies can be created using hybridoma technology in transgenic mouse models. A more contemporary platform known as antibody phage display allows for in vitro selection of human antibodies through isolation of high-affinity ligands. In this system, human immunoglobin gene libraries are displayed on the surface of bacteriophages and probed against antigens of interest to isolate a specific high-affinity antibody. Isolated mAbs can be further modified to improve functionality. Single-chain fragment variables (scFvs) consist of only the variable regions of the light and heavy chain and are connected through a peptide linker. In addition to scFvs, other formulations of the antibody heavy and light chain domains are possible. mAbs can further be conjugated to cytotoxic drugs for targeted delivery. Additional engineering to generate bispecific and trispecific molecules can also extend functionality ( Fig. 10.4 ). The following sections will review antibody-based therapies in further detail.
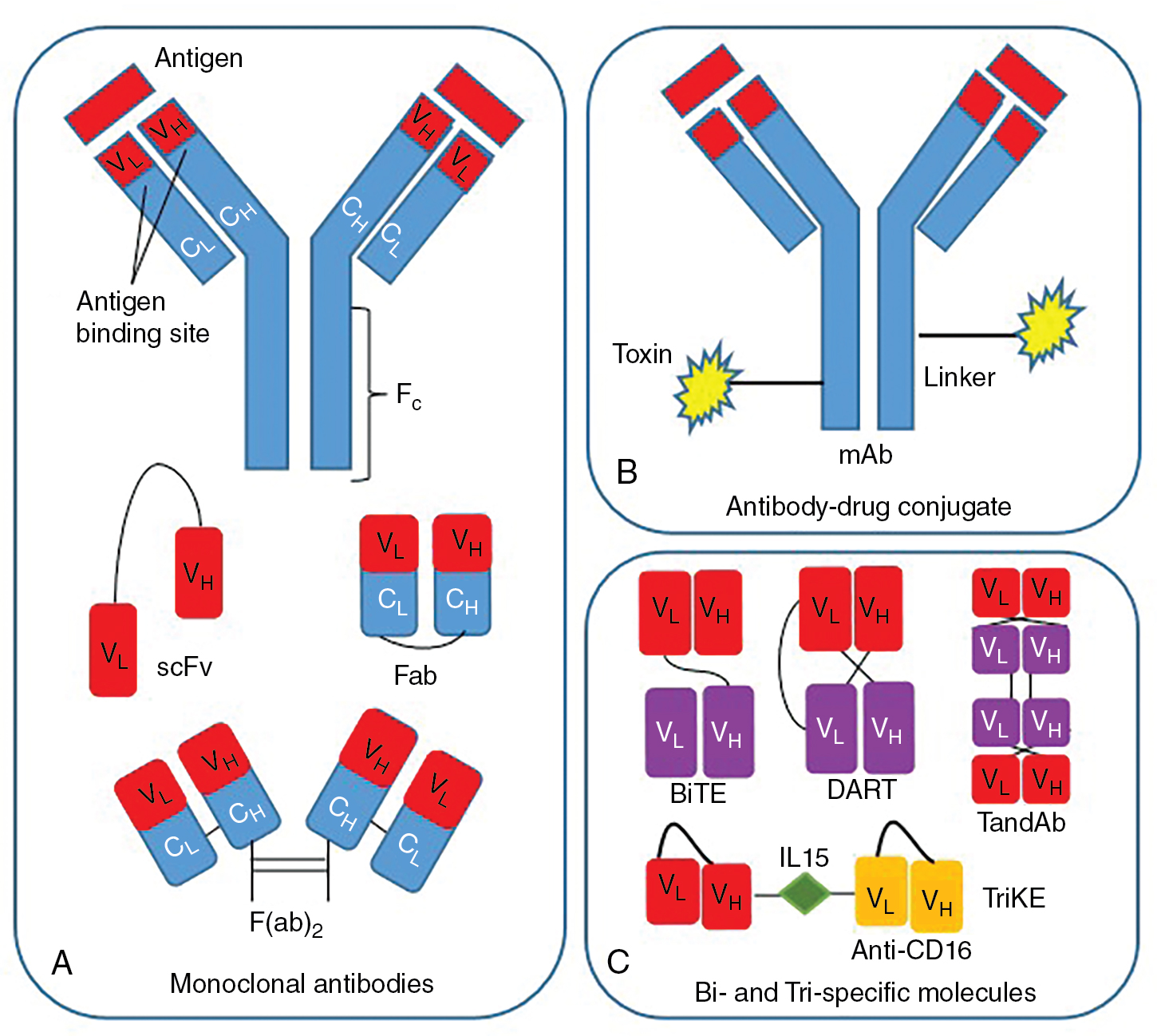
Monoclonal antibodies
Antibody-based therapies are among the most important and successful treatments for a rising number of cancer diagnoses. The general mechanism of action of most mAbs includes immune system redirection toward antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) for specific action against malignant cells. Many mAbs alter signal transduction via interaction with growth factor receptors at the target or downregulate expression or accessibility of key surface antigens. Their actions can further be combined with chemotherapy for synergistic effect. Of note, checkpoint inhibitors including ipilimumab (CTLA-4 inhibitor) and nivolumab and pembrolizumab (PD-1 inhibitors) represent a key category of monoclonal antibody therapy that has changed the treatment landscape for solid tumors, and are discussed in more detail in The role and application of checkpoint blockade . Table 10.1 provides a comprehensive overview of FDA-approved monoclonal antibodies for use in clinical practice. Notable monoclonal antibody therapies that have significantly advanced treatment of solid tumors and hematologic malignancies are highlighted in the following paragraphs.
| Name | Format | Target Antigen | Indication |
|---|---|---|---|
| Hematologic Malignancies | |||
| Alemtuzumab | Humanized IgG1 | CD52 | B-cell chronic lymphocytic leukemia |
| Daratumumab | Human IgG1 | CD38 | Multiple myeloma |
| Elotuzumab | Humanized IgG1 | SLAMF7 | Multiple myeloma |
| Isatuximab | Chimeric IgG1 | CD38 | Multiple myeloma |
| Mogamulizumab | Humanized IgG1 | CCR4 | Cutaneous T-cell lymphoma |
| Obinutuzumab | Humanized IgG2 | CD20 | Chronic lymphocytic leukemia, follicular lymphoma |
| Ofatumumab | Human IgG1 | CD20 | Chronic lymphocytic leukemia |
| Rituximab | Chimeric IgG1 | CD20 | B-cell lymphoma |
| Solid Tumors | |||
| Atezolizumab | Humanized IgG1 | PD-L1 | Bladder cancer, non-small cell lung cancer, triple-negative breast cancer, small cell lung cancer, hepatocellular carcinoma, melanoma |
| Avelumab | Human IgG1 | PD-L1 | Urothelial carcinoma, Merkel cell carcinoma, renal cell carcinoma |
| Bevacizumab | Humanized IgG1 | VEGF | Colorectal cancer, non-small cell lung cancer, renal cancer, glioblastoma, cervical cancer |
| Cemiplimab | Human IgG4 | PD-1 | Cutaneous squamous cell carcinoma, basal cell carcinoma, non-small cell lung cancer |
| Cetuximab | Chimeric IgG1 | EGFR | Colorectal cancer, head and neck squamous cell carcinoma |
| Dinutuximab | Chimeric IgG1 | GD2 | Neuroblastoma |
| Durvalumab | Human IgG1 | PD-L1 | Bladder cancer, non-small cell lung cancer, small cell lung cancer |
| Ipilimumab | Human IgG1 | CTLA-4 | Melanoma, renal cell carcinoma, colorectal cancer, hepatocellular carcinoma, non-small cell lung cancer, malignant pleural mesothelioma |
| Necitumumab | Human IgG1 | EGFR | Non-small cell lung cancer |
| Nivolumab | Human IgG4 | PD-1 | Melanoma, non-small cell lung cancer, renal cancer, classical Hodgkin lymphoma, head and neck cancer, urothelial carcinoma, colorectal cancer, hepatocellular carcinoma, esophageal carcinoma, malignant pleural mesothelioma, gastric cancer |
| Panitumumab | Human IgG2 | EGFR | Colorectal cancer |
| Pembrolizumab | Humanized IgG4 | PD-1 | Melanoma, triple-negative breast cancer, non-small cell lung cancer, head and neck squamous cell cancer, classical Hodgkin lymphoma, primary mediastinal B-cell lymphoma, urothelial carcinoma, microsatellite instability-high or mismatch repair–deficient solid tumors, colorectal cancer, gastroesophageal junction adenocarcinoma, cervical cancer, hepatocellular carcinoma, Merkel cell carcinoma, renal cell carcinoma, cutaneous squamous cell carcinoma |
| Pertuzumab | Humanized IgG1 | HER2 | Breast cancer |
| Ramucirumab | Human IgG1 | VEGFR2 | Gastric cancer, non-small cell lung cancer, colorectal cancer, hepatocellular carcinoma |
| Trastuzumab | Humanized IgG1 | HER2 | Breast cancer |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


