Summary of key facts
- •
Receptors of adaptive immune cells and the ensuing signaling networks are exquisitely specific and sensitive to new or altered antigens and thus poised to respond to tumors.
- •
The capacity of adaptive immune cells to traffic to sites of inflammation is programmed during initial priming and allows them to seek and destroy metastatic deposits throughout the body.
- •
The ability to form self-renewing memory cells that can reside in strategically important locations in the body can promote rapid responses to reexposure to antigen.
- •
Dendritic cells serve as conduits of both antigenic and inflammatory information between innate and adaptive immune systems.
- •
Dendritic cells exist in several maturation and differentiation states that have different capacities to acquire and present antigen in different costimulatory contexts.
- •
Cytokine production during antigen presentation by dendritic cells influences how T cells differentiate into effector and memory cell populations.
- •
Both T and B cells undergo metabolic alterations that provide the building blocks for their expansion and subsequent differentiation to either effector or memory populations.
- •
Chemokine receptors and integrins regulate the recruitment and trafficking of T and B cells both in secondary lymphoid organs and in inflamed peripheral tissues.
- •
Diversity in the differentiation state of helper CD4 + T cells promotes their ability to interact with and influence both innate immune populations such as macrophages and other adaptive immune cells including cytotoxic CD8 + T cells and antibody-producing B cells.
- •
The absence of inflammation within tumors limits dendritic cell activation and the quality of the ensuing immune response.
- •
Tumors and the tumor microenvironment contain molecules that are designed to limit adaptive immune responses and can limit immune-mediated destruction of tumors.
- •
Understanding the composition and function of adaptive immune receptors has allowed their development as therapeutic and diagnostic tools for cancer therapy.
- •
Biologic variations such as age and sex can have important influences on the quantity and quality of adaptive immune responses.
Introduction
Overwhelming evidence, both experimental and observational, indicates that cells of the adaptive immune system can limit the ability of tumors to grow and spread throughout the body. Mice with genetic lesions in molecules critical for the development or regulation of adaptive immune responses have a dramatically increased level of tumor formation and more advanced tumors than their wild-type counterparts. Transfer of lymphocytes from mice that have eradicated tumors can provide protection against tumor challenge in naïve, unexposed mice. The presence of lymphocytes, particularly T cells, within patients’ tumors is commonly a positive prognostic indicator.
To fulfill these functions, cells of the adaptive immune system must have mechanisms to sense and respond to subtle changes in antigens expressed by transformed cells and the ability to traffic to the site of tumor growth and perform functions that limit tumor growth. This is achieved by the transfer of antigen from transformed cells to diverse populations of professional antigen-presenting cells (APCs), primarily dendritic cells. These cells traffic from the periphery to lymph nodes, the hubs of interactions between APCs and lymphocytes, or acquire free antigen in the lymph node via lymphatic drainage and present antigen in the context of major histocompatability complex (MHC) molecules to T cells. Soluble antigen follows the lymphatics and diffuses into the subcapsular sinus where it can be recognized by B cells. Larger antigens are presented to B cells by trafficking dendritic cells, macrophages, or follicular dendritic cells.
If the T cells express a cognate receptor that can bind MHC–antigen complexes and the APC is expressing critical costimulatory molecules, T cells will expand, differentiate, and traffic back to the site of antigen origin. Once in the tissue, T cells reencounter antigen on MHC molecules expressed by tumor cells, or local APCs, and are triggered to perform effector functions such as cytolysis and cytokine release. These effector molecules can either directly eliminate tumor cells or induce innate immune cells within the tumor microenvironment to release further effector molecules and induce tumoricidal activity that constrains tumor growth. Some of the expanded T cells differentiate to long-lived memory cells that can reside in tissue, recirculate, or take up long-term residence in lymph nodes, poised to respond to further exposure to antigen.
B cells, once activated by their cognate antigen, differentiate either into extrafollicular antibody-producing cells, called plasmablasts ; traffic to germinal centers to become plasma cells whose antibody is modified by affinity maturation; or become long-lived memory B cells that are also capable of reexpanding into antibody-producing cells. The role of tumor antigen-specific antibodies in adaptive immune responses is not well defined. In addition, B cells can help promote nests of immune cells, known as tertiary lymph node structures , that can serve as local hubs that support and sustain both T- and B-cell responses to tumors.
Recognition of tumor antigens: Specificity engendered by the T-cell and B-cell receptors
A considerable advantage of immunotherapy compared with standard of care chemotherapies or radiation therapy is the exquisite specificity of the T-cell receptor (TCR) and the B-cell receptor (BCR)/antibodies for their cognate ligand. There are important differences in the nature of the ligand recognized by these two antigen receptors: the TCR binds to a complex of MHC and peptide derived from an independent protein, whereas antibodies, and thus the BCRs, bind to regions of free antigens without the involvement of other molecules ( Fig. 7.1 ). Further, TCRs generally recognize ligands that are formed from antigens that are produced either from within the cell (MHC class I) or from extracellular sources (MHC class II), whereas antibodies and the BCRs primarily recognize extracellular antigen (because antibodies cannot naturally penetrate the cell membrane). In this chapter we will explore the basis of this specificity and delineate the signaling events initiated by these receptors because this knowledge has a direct effect on not only understanding checkpoint inhibition therapy but also the differences in the iterations of chimeric antigen receptor (CAR)-T generations currently in therapy.

Structure of αβ T-cell receptor
The TCR consists of two polypeptide chains, α and β, that are composed of variable (V) and junction (J) domains for the α chain (VJ) and an additional diversity (D) region for the β chain (VDJ) ( Fig. 7.2 ). These chains result from somatic DNA recombination of germline genes during T-cell development in the thymus. Because gene rearrangement occurs at the somatic level, each T cell expresses thousands of copies of an identical single receptor after the recombination process is complete. This gene rearrangement is driven by recombinase-activating genes ( RAG1 and RAG2 ). The TCR α V locus comprises ∼80 gene segments and ∼60 J gene segments. The TCR β locus contains 52 V gene segments, 13 J gene segments, and 2 D segments. A single constant domain links the αβ TCR chains to signaling components of the CD3 complex that are responsible for initiating the signaling cascade within the T cell. Rearrangement of gene segments continues until the T cell receives a positive signal from self-MHC peptide complexes in the thymus or until the cell dies because of a lack of instructive signal from a successful rearrangement. The consequence of having two interacting chains that are derived from such a tremendous diversity of gene segments is the production of T cells each with a unique, highly specific TCRs capable of productively interacting with only a few MHC–peptide complexes. This notion is reinforced by evaluating crystal structures of the TCR, which show that the V region gene segments are clustered at the N-terminus of both the α and β chains, generating an antigen-binding domain that will contact the MHC–peptide complex.

A critical element in adaptive immunity is the ability to discriminate self-derived antigens from foreign or altered self-antigens. On the one hand, this is achieved with the incredible diversity and specificity of the TCR. However, an excessive T-cell pool is energetically demanding and raises the possibility of responding to self-antigens and the initiation of autoimmunity. One part of this puzzle is solved by survival signals in the thymus only being delivered to T cells whose TCR has an ability to engage the host MHC molecules. Those whose TCRs do not “match” the MHC of the host die by neglect. This process is referred to as positive selection . If the resulting TCR has too high an affinity for MHC-self peptide complexes, the bearing T cell will subsequently undergo activation-induced apoptosis, known as negative selection . , This process is required to limit autoreactive T cells from transiting to the periphery. Together, these selection events sculpt the T-cell repertoire such that it is focused on discerning alterations in the proteome of the host cells. Interestingly, some of the first identified tumor antigens were shown to be normal melanocyte proteins, suggesting that the selection process is not absolute.
Structure of the B-cell immunoglobulins
Significant similarities are present in the construction of antibodies and their receptor counterparts to the processes that occur with TCR construction. The BCR is composed of heavy and light chains, which together generate the antigen-binding domain ( Fig. 7.2 ). As with T cells, B-cell development and selection is a function of successful steps in the assembly of antigen receptor genes. Progenitor B cells begin by rearranging the heavy chain locus containing V, D, and J gene segments. D-J recombination occurs first, followed by recombination with V genes, and finally the constant (C) region is added. In humans, the heavy chain locus contains 38 to 46 V gene segments, 23 D gene segments, 6 J segments, and 9 C segments. Two light chains, k and l, are available to pair with the heavy chain, and each of these has a high number (∼29–38) of V genes and 5 J genes but no D genes. As with the TCR, this diversity results in a tremendous number of possible sequences encoded in the antigen-binding region and thus the repertoire of antigens that can be bound. Further variability is generated in antibody segments by two additional steps: (1) imprecise joining of gene segments, resulting in junctional diversity, and (2) DNA repair enzymes and terminal deoxynucleotidyl transferase activity removing and adding nucleotides randomly to the single-strand ends.
Whereas the antigen binding fragment (Fab) is responsible for the neutralizing and absorptive activities of antibodies, for immunoglobulins, the addition of different constant regions (M, D, G, E, and A) to the heavy chain, thus defining their isotype , imbues different functional activity on the molecule. These include different efficiencies in activating complement, binding to Fc receptors, and ability to cross the placenta. Immunoglobulin (Ig) D and IgM are translated from the same pre-mRNA transcript and are coexpressed on the surface of naïve, mature B cells. These heavy chains undergo instructional class-switching after first engagement with antigen, under the guidance of T cells. Importantly, transmembrane (B-cell receptor) and secreted (antibody) forms of these immunoglobulins are generated from the same mRNA transcript by differential splicing of mRNA to replace exons encoding a membrane anchor with exons encoding secretion sequences. These concepts are more thoroughly described in Chapter 2 . The secreted forms of antibodies have been extensively exploited in therapeutic settings, particularly once the development of monoclonal antibody hybridoma technology (where an immortalized B cell secretes large quantities of a single antibody) allowed the production of large quantities of homogeneous, highly defined antibody product. Some of the uses of monoclonal antibodies in cancer therapy are described in Table 7.1 .
| Type | Intent | Example |
|---|---|---|
| Antagonist | Blocks receptor–ligand interactions | Impeding checkpoint molecules; e.g., PD-1 or CD73; CSF-1 |
| Agonist | Cross-linking activating receptors | Stimulating costimulatory molecules; e.g., CD134 (OX40), CD40 |
| Antibody–drug conjugate | Tumoricidal activity | HER2-neu; ado-trastuzumab emtansine (T-DM1) |
| Imaging | Detection of metastases | Indium-111 satumomab, colorectal and ovarian cancer |
| Antigen capture | Monitoring tumor burden | CA-125; PSA |
| Antigen capture | Immune function assays | Cytokine ELISA; ELISPOTs |
Understanding the proteomic and genetic makeup of antibodies has allowed the further development of this resource beyond the initial hybridoma approaches to those that include isolation and expression of antibodies from individual B-cell clones and approaches that limit the inherent immunogenicity of murine proteins injected into humans. Note that exploration of synthetic TCRs as potential therapeutics, , targeting MHC–peptide complexes presented at the surface of tumor cells, is an ongoing and expanding field of research.
Antigen receptor signaling
Signaling from T-cell receptor
For naïve T cells to proliferate and differentiate after engagement of the TCR by MHC–peptide complexes presented by dendritic cells (DCs), or for effector T cells to perform their functions upon reencountering their cognate antigen, signals must by propagated within the T cell, generally via the induction of enzymatic activity. Many of the common principles of receptor signaling were originally determined in the study of T cells. Knowledge of the mechanistic basis of TCR signaling has been critically important for understanding how checkpoint molecules such as Cytotoxic T lymphocyte Antigen -4 (CTLA-4) and Programmed Death 1 (PD1) work, and how the construction of more effective chimeric antigen receptor T cells (CAR-T) can be achieved.
Antigen-binding αβ T-cell receptor chains have no signaling capacity on their own. Rather, they associate with other molecules at the cell surface, CD3γ, CD3δ, CD3ε, and a ζ chain, which together form the CD3 complex (see Fig. 7.3 ). , The CD3 molecules are composed of immunoglobulin-like structures that have extracellular domains, and the ζ chain is primarily composed of a longer intracellular domain with only a short extracellular addition to the transmembrane domain. Critical to the ability to propagate signals, the intracellular domains of the CD3 proteins and the ζ protein contain immunoreceptor tyrosine-based activation motifs (ITAMs). , These ITAMs become phosphorylated by protein tyrosine kinases (PTKs) when the receptor complex engages its ligand. PTKs exist in two forms: as either intrinsic components of receptors (receptor tyrosine kinases [RTKs], such as the Fms-like tyrosine kinase 3 ligand (FLT3L) receptor) or as non-RTKs. An example of non-RTKs would be Janus Kinases (JAK) associated with cytokine receptors or Lymphocyte-specific protein tyrosine kinase (Lck) associated with the TCR. Non-RTKs are either constitutively associated with their receptors or are recruited after ligand binding. RTKs often multimerize to activate or are kept in inactive states by posttranslational modifications, as is the case with Lck.
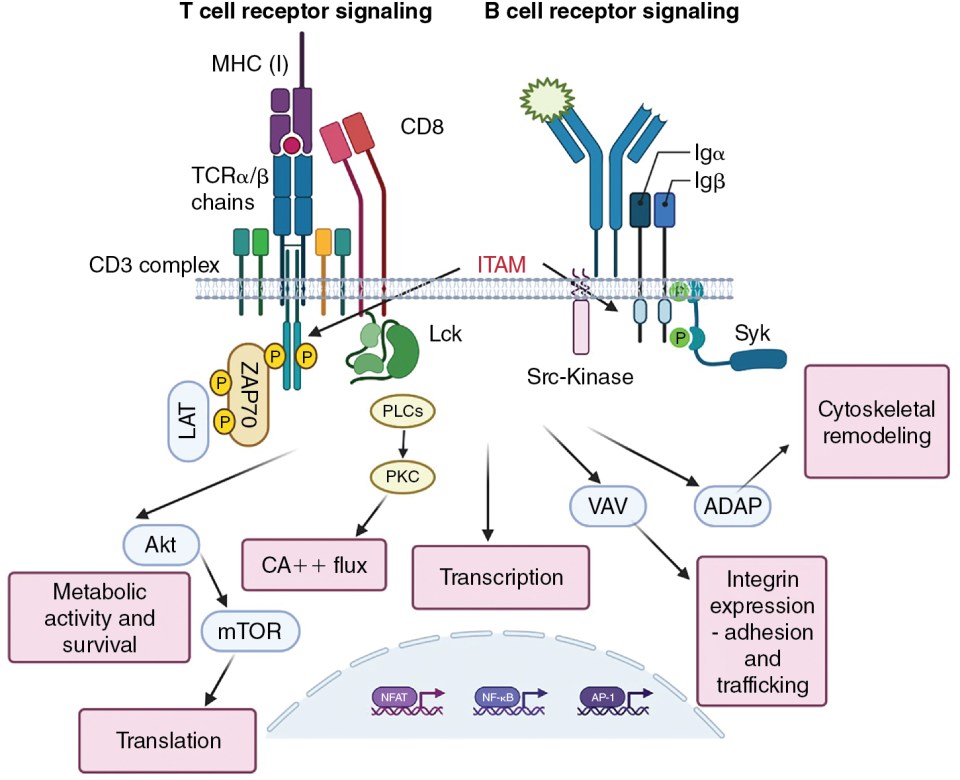
Initial signals
Charge interactions lead to the assembly of the TCR complex, which occurs in the endoplasmic reticulum and is required for export to the plasma membrane. ITAMs found in the invariant chains of the TCR drive phosphotyrosine signaling. In the case of the TCR, this function is performed in both CD4 + and CD8 + T cells by Lck. In naïve T cells, Lck is normally kept in a nonfunctional confirmation by autophosphorylation and phosphorylation by Csk ; it is released to function in the activation of TCRs by the CD45 phosphatase. The initial phosphorylation of CD3 ITAMs is performed by Lck, allowing the recruitment of Src homology 2 (SH2)-containing scaffold proteins, including Linker for Activation of T cells (LAT) and SH2 domain containing leukocyte protein of 76kDa (SLP76) to the TCR complex (see Fig. 7.3 ). Antigen receptor ITAMs have two phosphorylation motifs, and the signaling intermediary ZAP70 has tandem SH2 domains that match these phosphorylation sites. This stoichiometry enhances the specificity and interactivity of ZAP70 with the ε and ζ chains of the TCR. It is worth remembering that the ability of the TCR to recognize antigen must be very sensitive (because of the low copy numbers of MHC–peptide on APCs) and very specific (must not respond to similar ligands). , The arrangement and interactivity of the TCR signaling complex likely contributes to making it relatively difficult to activate, limiting nonspecific activation, but the presence of multiple enzymes also strongly enhances the signal once activated. It is currently unclear how antigen binding affects the stoichiometry/confirmation of the TCR complex to initiate signaling, and clustering of TCRs is difficult when antigen (i.e., infrequent MHC–peptide complexes on APCs) density is low. Microclusters of TCRs may form, changing the apparent concentration of TCRs and the ratio of activating and inactivating enzymes. ,
The intensity, and thus effectiveness, of the signal varies with the affinity of the ligand (interestingly, the TCR has relatively low affinity for MHC–peptide complexes compared with antibody/BCR for their antigen; it is thought that this is necessary to allow sustained T-cell activation), the concentration of the ligand, the activity of phosphatases, and the concentration and availability of intracellular signaling components. Thus TCR signaling can by tunable and dynamic, and the signal strength has qualitative effects on downstream T-cell activity. The system is reset by a combination of phosphatases and ubiquitination and degradation of recycling receptors.
The signaling complex formed after initial TCR engagement is responsible for activating the enzymes that work downstream of TCR signaling to promote the functional activities of T cells ( Fig. 7.3 ). These include Phospholipase C γ (PLCγ), which activates transcription factors via the production of Inositol trisphospate (IP3) and the initiation of Ca ++ flux, which in turn serves as critical amplifier of post-TCR signals. The transcription factors for full T-cell activation include nuclear factor of activated T cells (NFAT), activator protein 1 (AP-1) (for interleukin [IL]-2 production), and nuclear factor kappa B (NF-κB). , Activation of protein kinase B (AKT) (a serine/threonine kinase) promotes metabolic changes and initiates mammalian target of rapamycin (mTOR) activation, which is critical for protein translation. Further pathways of activation include Vav, which controls cytoskeletal remodeling necessary, and adhesion and degranulation promoting adaptor protein (ADAP), which instigates the expression of integrins needed for cellular adhesion and trafficking. This remodeling drives the accumulation of the appropriate molecules into the contact site between the T cell and the antigen-presenting cell, known as the supramolecular activation complex (SMAC).
Many forms of severe combined immunodeficiency (SCID) result from defects in Lck, ZAP70, and other RTK involved in regulating T-cell activation.
B-cell receptor signaling
The BCR serves both as a signaling receptor, inducing the propagation of events that lead to functional changes within the B cell, and as an antigen-binding receptor for the purposes of antigen processing and presentation. Though in some instances, B cells can respond to antigen directly (T-independent antigens), these antigens tend to be associated with pathogenic infections and their allied adjuvant effects. Most B-cell responses require support of T cells, and the antigenic targets of these responses are referred to as T cell-dependent. Many aspects of BCR signaling are analogous to TCR signaling, because the antigen-binding domains of the BCR are associated with ITAM-containing components (Igα and Igβ) that transmute the signal. In place of Lck, B cells use the protein tyrosine kinases Fyn, B lymphoycte kinase (Blk) and Lyn to phosphorylate ITAMs after BCR multimerization. Spleen tyrosine kinase (Syk) serves as the ZAP-70 equivalent in B cells and it contains two SH2 domains that allow recruitment to the phosphorylated BCR. In each case, the primary proliferative signal from the BCR is communicated via PI-3 kinase-mediated activation of RAS/MAPK (mitogen activated protein kinase) pathway, inducing the activation of the transcription factors AP-1 and NFAT. The coreceptor functions of CD4 and CD8, and to a certain extent CD28 costimulation, are replaced in B cells by the CD19–CD21–CD81 complex. CD21 binds complement CD3dg–decorated antigen and the ensuing activation results in the activation of Src family tyrosine kinase. This promotes further Syk activation and the recruitment of the scaffolding protein SLP-65 (a LAT-SLP76 equivalent) to the BCR, and phosphorylation of CD19 permits the recruitment of phosphatidylinositol 3-kinase (PI3K). As a consequence of these activation events, the B cell can initiate remodeling events very similar to that seen in T cells, with phosphatidylinositol (3,4,5)-trisphosphase (PIP 3 )/3-phosphoinositide-dependent kinase 1 (PDK1) leading to the activation of AKT, Bruton’s tyrosine kinase (BtK) leading to the activation of phospholipase C-gamma (PLC-γ) to make diacylglyceride (DAG) and inositol trisphosphate (IP3), and Vav activating Wiskott-Aldrich Syndrom Protein (WASP) and actin polymerization that precedes synapse formation that is necessary for antigen uptake and subsequent engagement of T helper cells. Targeting BtK is a therapeutic option for some B-cell malignancies.
Costimulation
CD28-signal 2
Antigen receptor binding (Signal 1) is insufficient to drive the full activation of naïve T and B cells. A second, conditioning, signal is needed to promote and sustain the activation and cycling of lymphocytes. In the case of T cells, in addition to TCR recognition of Ag/MHC complexes expressed on an APCs, such as a virus-infected dendritic cell, costimulatory signaling occurs via interaction between the costimulatory molecule CD28 and B7.1 or B7.2 (CD80/CD86), , if the APC expresses them. This leads to induction of both the high-affinity IL-2 receptor (IL-2Rα) and IL-2, which provides sufficient signals for proliferation, and the development of effector (cytotoxic) functions such as the generation of cytolytic granules. Recognition of the Ag–MHC complex is sufficient to drive the T cell from G0 into the G1 phase of the cell cycle but not through S phase (DNA replication). As such, the production of IL-2 can almost be considered a go–no go signal for T cells. Because Lck can also phosphorylate CD28 (if it is in the SMAC), , it can lead to the recruitment of growth factor receptor-bound protein 2 (Grb2), ultimately leading to the generation of more PIP 3 . This has the effect of supporting the signaling modules described previously: PLCγ and downstream transcription factors, AKT and Vav, and the stabilization of IL-2 mRNA. CD28 stimulation is considered as Signal 2 in T-cell activation.
In the absence of Signal 2, T cells enter a state of unresponsiveness termed anergy . Anergy is not passive but is sustained by the expression of ubiquitin ligases such as gene related to anergy in lymphocytes (GRAIL) and c-Casitas B-lineaage (c-CBL) that actively degrade components of the TCR signaling process, limiting T-cell activation. , As might be anticipated, high doses of IL-2 can overcome the induction of anergy, likely by binding to the low-affinity IL-2βγ chain receptor. Anergy is induced in T cells when APCs are insufficiently activated (because of limited inflammation) to upregulate CD80 and/or CD86 but do present MHC–peptide complexes. This can occur in situations of peripheral tolerance (when new antigens arise as a function of developmental stages such as puberty) or in the case of slow-growing tumors. Importantly, the expression of CTLA-4, which serves as a competitor to CD28 on T cells for binding with CD80/CD86, , mimics the effects of encountering antigen without strong costimulation. Approaches that impede CTLA-4 have had some beneficial effects in several cancer entities. Other costimulatory molecules include IgSF members such as inducible costimulator (ICOS) and members of the tumor necrosis factor receptor superfamily (TNFRSF), such as CD27, OX40, and 41BB. Stimulation of TNF superfamily molecules promotes the expression and activity of c-Jun N-terminal kinase (JNK) via their regulation of tumor necrosis factor receptor-associated factor (TRAF) molecules and thus NF-κB. TNFRSF stimulation is primarily associated with T-cell survival, although some aspects of metabolic activity and memory cell differentiation have been attributed to these costimulatory molecules. TNFRSF receptors are often expressed 2 to 3 days after initial activation of DCs, providing a sustaining signal to activated T cells.
For B cells, the source of costimulation differs between the two types of antigens. T cell–independent BCR signals require support from the CD91–CD21–CD81 complex, which engages complement-bound antigens. This results in SH2-mediated enhancement of BCR signaling and the engagement of scaffolding signaling complexes that promote and multiply initial BCR signals. Further T cell–independent costimulation is supported by sensing additional pathogen-associated molecular patterns (PAMPs) from invading pathogens. These provide a MyD88-dependent activation of the NF-κB pathway that supports B-cell activation, analogous to the role of NF-κB in T cells.
The BCR serves both as a signaling receptor, inducing the propagation of events that lead to functional changes within the B cell, and also as a mechanism for internalizing antigen for presentation on MHC class II molecules, both turning the B cell into a professional antigen-presenting cell (APC). This ability to process and present antigen on MHC class II molecules allows B cells to interact with antigen-specific CD4 + T cells (in this case, T follicular helper cells [Tfh] located in the lymph nodes), which in turn support B cells by providing cytokines and receptor ligands that support B-cell proliferation and differentiation into either antibody-secreting cells or memory B cells (T cell–dependent processes). Tfh signals include CD40L, which provides survival advantages via the noncanonical NF-kB pathway (mediated by NF-kB-inducing kinase [NIK]) and the induction of the survival factor BCL2, and IL-21, which signals via signal transducer and activator of transcription 3 (STAT3) to promote proliferation and differentiation into memory and plasma cells. B cell–activating factor (BAFF; also known as B lymphocyte stimulator, BLyS), a member of the TNF superfamily, provides additional survival signals for maturing B cells.
Exhaustion
T-cell responses to cancer will be limited by the lack of costimulation during antigen presentation, leading to anergy, as described previously. In addition to anergy, T-cell responses within the tumor microenvironment are often curtailed because of the effect of chronic antigen presentation. , In this context, the consistent presence of antigen prevents the programmed acquisition of memory T-cell characteristics and the loss of pluripotent stemness. Aligned with this is a reduced expression of cytokines, reduced killing capacity, and limited proliferation. As with anergy, this altered state is driven by changes in transcriptional activity (e.g., the ratios of T-box expressed in T cells (Tbet): Eomesodermin (Eomes), T cell factor 1 (TCF1): Thymocyte Selection Associated High Mobility Group Box (TOX), , and B-cell lymphoma 6 (Bcl6): B lymphocyte-induced maturation protein-1 (BLIMP-1) , ) and the expression of a wide array of checkpoint inhibitor molecules, including Programmed Death-1 (PD-1), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) ( Table 7.2 ), whose ligands are commonly expressed in the tumor microenvironment (TME). The need for such an extensive network of counterstimulatory molecules is unclear. Transcriptional changes in exhausted T cells are quite distinct from those found in anergic T cells. Mechanistically, many of these molecules work by recruiting phosphatases (e.g., SHP2 in the case of PD1) that disrupt signal transduction pathways, thus attenuating signals from the TCR. Exhaustion is accompanied by metabolic alterations and, with time, epigenetic repression of key effector cell activities. Extensive studies in experimental models and in clinical trials have demonstrated the efficacy of targeting these checkpoint molecules for cancer immunotherapy. Correlates of responsiveness in patients include the presence of T cells within the patient’s TME and tumor mutational burden (TMB), which reflects the array of putative tumor antigens available for T cells to target. , In some instances, high doses of IL-2 or IL-7 can revert exhausted phenotypes.
| Receptor | Potential Ligand | Possible Mechanisms of Action |
|---|---|---|
| CTLA-4 | CD80/CD86 | Competes against CD28 costimulation |
| PD1 | PDL1; PDL2 receptors | Recruits SHP-2 phosphatase to SMAC via ITIM; limits TCR and CD28 signals |
| Unknown | B7H3 (CD276) | Unclear; T-cell proliferation reduced |
| LAG3 | MHC class II; L-SECtin; Gal3; FGL-1; α-syn | Diminishes TCR signaling |
| TIM3 (CD366) | GAL9; phosphatidylserine; HMGB1 | Impedes DC activation by DAMPs; induces apoptosis of Th1 |
| TIGIT | CD112/CD155 | Competes with CD226 costimulation |
| BTLA | HVEM | SHP-1 and SHP-2 recruitment |
| CD160 | HVEM | Lower IL-2 mRNA; CD3ζ phosphorylation |
| PSGL-1; VSIG3 | VISTA (B7-H5) | Induces FOXP3 |
| 2B4 (CD244; SLAMF4) | CD48 (SLAMF2) | ITSM-mediated SHIP, SHP-1, SHP-2 recruitment; CsK recruitment |
| Unknown | B7-H3 | Limits IFN-γ production; indirect effects by modulating MDSC |
| Unknown | B7-H4 | Arrests cell cycle |
Evidence for a similar exhaustion phenotype in B cells is mostly derived from studies of chronic/latent pathogen infections. B-cell exhaustion is also characterized by the expression of ITIM-containing inhibitory receptors, such as Fc receptor-like 4 (FCRL4) and sialic acid–binding Ig-like lectin 6 (Siglec-6) and downregulation of proliferative activity and BCR signaling. The presence and function of exhausted B cells in the tumor microenvironment has not been well documented.
Senescence
As with exhaustion, cellular senescence is a state of limited responsiveness that arises because of chronic antigen engagement and with aging (see T-cell subsets in cancer immunity ). Senescent T cells are characterized as having reduced telomere lengths and increased DNA damage, perhaps reflective of the combination of extensive replication and the age of the host. The DNA damage response found in senescent T cells can be induced both by tumor cells themselves as well as regulatory T cells found within the tumor microenvironment. Further characteristics that demark senescent T cells from exhausted include the absence of the costimulatory molecules CD27 and CD28, upregulation of molecules associated with terminal differentiation killer cell lectin like Rreceptor G1 (KLRG1; CD57), and the extensive production of proinflammatory and antiinflammatory cytokines. Some checkpoint molecules associated with exhaustion can also be expressed (e.g., TIM3, TIGIT) by senescent T cells. Proliferation is compromised by lesions in the proteins that are normally activated by TCR signaling, limiting the accumulation of phosphorylated signal transduction complexes.
Metabolic adaptations used by lymphocytes
T-cell metabolic programming
Considering the rapid proliferation by lymphocytes and the engrailment of effector activities, significant modifications occur in their metabolic activity to support these changes. At the simplest conceptual level, naïve and memory T cells are considered metabolically quiescent, or anabolic, whereas effector T cells use catabolic processes to convert glucose into the building blocks of protein, nucleotide, and lipid synthesis necessary for rapid proliferation ( Fig. 7.4 ). Quiescent T cells uptake glucose at modest levels and use oxidative phosphorylation to generate adenosine 5’-triphosphate (ATP). Quiescent T cells rely on fatty acid oxidation to produce ATP and nicotinamide adenine dinucleotide (NAD) + hydrogen (H) (NADH) in the mitochondria via the electron transport chain. Importantly, inhibition of fatty acid oxidation impairs memory T-cell formation, persistence, and function. With the engagement of the TCR, initial proliferation is dependent on oxidative phosphorylation, but then T cells rapidly increase their glycolytic activity. , In T cells, the induction of MYC results in the elevated expression of glucose transporters and the enzymes of the glycolytic pathway. Because the production of ATP is substantially lower via glycolysis compared with oxidative phosphorylation, at first this seems counterintuitive. However, the amount of glucose uptake increases considerably, compensating for the potential shortfall. Additionally, the metabolic kinetics of boosting mitochondrial production to enhance oxidative phosphorylation, compared with augmenting glycolytic enzyme production, factor into this. Critically, in addition to ATP production, NAD + is reduced to NADH, the cofactor for a plethora of enzymes, during glycolysis. The intermediaries of glycolysis are also used by the pentose phosphate pathway and other metabolic processes for the synthesis of nucleotides (e.g., glucose-6-phosphate), amino acids (e.g., 3-phosphoglycerate), and pyruvate/citrate and acetyl co-A, which are foundational for fatty acid and lipid synthesis. Importantly, impeding glycolysis shuts down both the latter stages of T-cell proliferation and effector function activity. , In the latter case, data suggest that enzymes involved in the glycolytic pathway can serve as translational inhibitors of cytokine production when not engaged in glycolysis. TCR engagement also activates amino acid uptake for incorporation into protein synthesis, glutamine metabolism, and fatty acid synthesis. It has been argued that competition between tumors (which commonly use glycolysis) and T cells for glucose is a major metabolic barrier for successful T-cell function in the tumor microenvironment. ,

In contrast to effector T cell subsets, active regulatory T cells (Tregs) have been shown to use fatty acid oxidation as their primary metabolic activity. Blocking fatty acid oxidation limits Treg suppressive activity and promotes tumor control, implicating fatty acid oxidation as an active repressor of effector activity. Further, engagement of PD-1 increases the expression of carnitine palmitoyltransferase 1A (CPT1A), which is responsible for fatty acid transport into mitochondria, and higher levels of fatty acid oxidation, linking checkpoint inhibition with T-cell metabolism. It should also be noted that a variety of immunosuppressive mechanisms active in the tumor microenvironment involve the degradation, and thus availability, of nutrients necessary for T-cell function. These include arginase which depletes arginine, and indoleamine-2-3-dioxygenase (IDO), which catabolizes tryptophan. Glucose availability is not the sole metabolic regulator of T-cell function in the tumor microenvironment; oxygen deprivation due to poor angiogenesis has also been implicated as a metabolic barrier for T-cell activity against tumors ( Table 7.3 ).
| Metabolic Constraint | Example | Target |
|---|---|---|
| Fuel | Glucose | Glut receptors |
| Glutamine | Glutaminase | |
| Amino acid depletion | Tryptophan; arginine; cysteine | IDO; arginase |
| Immunosuppressive molecule production | Adenosine | CD39; CD73; A2AR |
| Lactate | MCT1; MCT4 | |
| Hypoxia | Angiogenesis; PDL1 upregulation | VEGF |
B-cell metabolic programming
Similar to T cells, naïve and memory B cells are maintained in a state of quiescence, primarily using oxidative phosphorylation to provide energy. Upon antigen receptor stimulation, B cells must undergo metabolic alterations to generate the secretory machinery that will be needed by plasma cells to produce copious quantities of antibodies. BCR-stimulated B cells undergo metabolic adaptations that are highly similar to those of T cells, increasing glucose uptake, glycolytic activity, and the production of pyruvate for consumption in mitochondria via the TCA cycle. With B cells, the antigen receptor initiates glucose transporter protein type 1 (GLUT1) upregulation in a PI-3K-dependent manner and glycolysis via phospholipase C-γ2 (PLC-γ2)- mediated activation of protein kinase-C β (PKCβ). Consistent with T cells, c-Myc promotes glycolysis, glutaminolysis, and amino acid uptake by B cells and is accompanied by the upregulation of a plethora of solute transporter proteins that enhance acquisition of metabolites from the extracellular environment. Indeed, the upregulation of CD98hc (Slc3a2) has been shown to be critical for B-cell proliferation. Hypoxia-inducible factor 1-alpha (HIF-1α) also contributes to the transcription of genes involved in solute transport and glycolysis. In contrast to T cells, nutrient sensing by B cells (and thus metabolic regulation) is apparently independent of AMP-activated protein kinase (AMPK) and the increase in glycolysis that occurs with antigen receptor stimulation is more coordinated with glucose oxidation. Notably, plasma cells also engage de novo lipogenesis to augment sufficient phospholipids to promote antibody secretion, and the hexosamine pathway repurposes glucose to allow antibody glycosylation.
Tumor antigen presentation: Dendritic cells
Role in the lymph node
In order for T-cell responses to be initiated, naive T cells bearing cognate TCRs, as described above, must encounter an MHC–peptide complex that has sufficient affinity to trigger signal transduction in the surveilling T cell. In addition to this first signal (Signal 1), the context in which antigen presentation occurs is critical to generate a productive T-cell response (the subsequent proliferation and differentiation of the stimulated T cell). The qualitative nature of antigen presentation is referred to as costimulation , or Signal 2. To successfully stimulate a primary T-cell response, Signal 1 and Signal 2 must be achieved simultaneously, meaning that the antigen-presenting cell responsible for initiating this response must be appropriately supplied with antigen and also educated, or activated, to become a professional APC. Dendritic cells are the predominant cell with these capabilities and thus play a critical role in eliciting immunity to tumors, whether, for example, as part of a cancer vaccine therapy or cells that participate in responses after initial tumor destruction elicited by standard of care therapies or targeted therapies. Each of these stages will be explained later.
Antigen presentation
The fundamental basis for activation of T cells is the presence of cognate MHC–peptide complexes on the cell surface. For CD4 + T helper cells, which will be explored in more detail later, this requires MHC class II molecules. In contrast, antigen presentation to cytotoxic CD8 + T cells requires MHC class I molecules. These two molecules are used because they generally acquire antigen from different cellular compartments. In most instances, MHC class I molecules acquire the peptides they present at the cell surface from proteins that have been expressed in the host cell (e.g., normal host proteins, proteins that contain alterations as a function of the transformed nature of the cell, or proteins that result from pathogenic infection). To access MHC class I molecules, protein products of translation are degraded in the cytoplasm by the proteosome and transported into the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) molecules where they can meet empty, recently translated MHC I molecules ( Fig. 7.5 ). Various editing proteins trim and enhance the binding of peptides to the MHC. Most peptides bound to MHC class I molecules are 8 to 10 amino acids in length. Subsequently, folding with β2-microglobulin induces the release of the complete MHC–peptide complex to the cell surface. In contrast, nascent MHC class II molecules move from the ER to endosomal vesicles containing antigen that has been acquired, by either endocytosis or phagocytosis, from the extracellular environment. As antigen-containing endosomes move inward, proteolytic enzymes such as cathepsins are responsible for digesting the antigen cargo, resulting in polypeptides that can, with the help of chaperone molecules such as human leukocyte antigen (HLA)-DM and HLA-DO, bind in the MHC class II binding cleft ( Fig. 7.5 ). Peptides derived from endogenous proteins are less commonly found on MHC class II molecules because the coexpressed invariant chain blocks peptide access until late endosomes, where it is also cleaved, allowing peptide binding to the MHC class II molecule. The different antigenic requirements for MHC molecules play a considerable role in the design of subunit cancer vaccines. Short, precise peptides are used for MHC class I binding and CD8 + T-cell responses, and longer peptides can be used to elicit MHC class II binding and CD4 + T-cell responses.

Related posts:
 Development and structure of the lymphoid system
The chemistry, structure, and function of immunoglobulins
The role of the complement system in cancer etiology and management
The major clinical components of cancer immunotherapy (modulating cell-mediated immune mechanisms)
The microbiome and cancer immunotherapy
The clinical application of immuno-therapeutics
Development and structure of the lymphoid system
The chemistry, structure, and function of immunoglobulins
The role of the complement system in cancer etiology and management
The major clinical components of cancer immunotherapy (modulating cell-mediated immune mechanisms)
The microbiome and cancer immunotherapy
The clinical application of immuno-therapeutics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


