Summary of key facts
- •
The complement system is a key component of the innate immune system, responsible for facilitating immune defense mechanisms, regulating inflammation, and maintaining tissue homeostasis.
- •
The complement system is a proteolytic cascade, comprising more than 50 highly regulated soluble proteins and membrane-bound receptors. The cascade is initiated by recognition of pathogen-associated patterns (PAMPs), damage-associated molecular patterns (DAMPs), and binding to immune complexes and proceeds via three distinct pathways: the classical, the lectin, and the alternative pathways.
- •
Complement activation results in production of important immune mediators, including C3a, C3b, C5a, and C5b-9, which mediate innate immune responses against foreign invaders, attraction of immune effector cells, phagocytosis, and formation of the membrane attack complex (MAC) resulting in cell lysis. This process is tightly controlled by a range of soluble and membrane-bound regulatory proteins.
- •
Though essential to immune function, dysregulated complement activation contributes to a diverse range of disease pathologies, including atypical hemolytic uremic syndrome, C3 glomerulopathies, age-related macular degeneration, rheumatoid arthritis, sepsis, atherosclerosis, ischemia-reperfusion injury, and cancer.
- •
Complement-targeting drugs have been FDA-approved for mostly orphan conditions such as paroxysmal nocturnal hemoglobinuria, yet several more are in clinical trials or under development.
- •
Complement is activated in response to cancer cells, and complement activation products are deposited within tumor tissue. Bioinformatics analysis indicates a negative correlation between complement signaling pathways and prognosis in many cancer types.
- •
Complement mediators including C3a, C5b, and sublytic C5b-9 have been implicated for roles in promoting tumor growth and metastasis.
- •
Research in preclinical models suggests the therapeutic potential of complement-targeting drugs, either alone or in combination with current cancer therapeutics.
Introduction
The complement system is critical to proper immune function, but inappropriate or excessive complement activation contributes to many pathologic inflammatory conditions including cancer. Traditionally regarded as contributing to the antitumor response through complement-dependent cytotoxicity (CDC), there is growing evidence that complement activation products including C3a, C5a, and C5b-9 can also promote tumor growth. Indeed, complement proteins have been shown to regulate primary tumor growth and metastasis, indirectly via the antitumor immune response, angiogenesis, and formation of the premetastatic niche and directly by promoting tumor cell proliferation and migration. The availability of complement-targeting drugs, with more in the developmental pipeline, suggests the potential for novel immunotherapeutic strategies to target both innate and adaptive immunity and boost the antitumor response.
Complement activation pathways
A key component of innate immunity, the complement system forms the first line of defense, aiding in the elimination of pathogens and damaged cells. The complement system is a proteolytic cascade, comprising more than 50 highly regulated soluble proteins and membrane-bound receptors. Complement activation elicits a range of proinflammatory effects, including increased vascular permeability, modulation of cytokine release, recruitment of innate immune cells such as neutrophils and macrophages to damaged tissues, enhanced phagocytosis, and lysis of pathogens and damaged cells. , Although it has been traditionally regarded as a mediator of innate immune activities, the complement system also contributes to efficient adaptive immune responses.
The complement cascade is activated in response to danger signals (damage- or pathogen-associated molecular patterns; DAMPs and PAMPs) via three pathways ( Fig. 3.1 ), depending on the stimuli. The classical pathway is activated via interaction of antigen–antibody complexes with the multimeric collectin C1q leading to conformational changes in the C1q molecule and complex formation with serine proteases C1r and C1s. The lectin pathway is initiated by the binding of mannan-binding lectin (MBL), ficolins, and other pattern recognition molecules that recognize aberrant carbohydrates on the surfaces of pathogens and damaged or necrotic cells, allowing for the recruitment of MBL-associated serine proteases (MASPs)-1 and -2. The alternative pathway is triggered by interaction with foreign antigens on pathogen surfaces. The activation of classical and lectin pathways, leading to formation of the C1qrs complex and activation of MASP-2, respectively, results in proteolytic cleavage of C4 into C4a and C4b and then C2 into C2a and C2b. C4b then binds to C2b, generating the classical/lectin C3 convertase (C4b2b complex). , The alternative pathway, however, is in a constant state of low-level activation (tickover), in which spontaneous hydrolysis of a labile thioester bond converts C3 to a bioactive form C3(H 2 O) in the fluid phase. This pathway proceeds directly through C3 cleavage to generate an alternative pathway C3 convertase (C3bBb), thus allowing an immediate response to microbial challenge.

Once generated, C3 convertases cleave C3 to produce the important effector molecules anaphylatoxin C3a and opsonin C3b. Formation of C3b also enables binding of the protease factor B (FB). The resulting proconvertase (C3bB) is quickly transformed by factor D (CFD) into an active C3 convertase (C3bBb) that by itself can cleave more C3 into C3b, thereby creating an amplification loop for C3b deposition. C3 convertases from the classical or alternative pathways also bind to C3b to generate C5 convertases (C4b2b3b and C3bBb3b, respectively). These in turn cleave the downstream component C5 to generate anaphylatoxin C5a and C5b, which initiates formation of the membrane attack complex. These products of complement pathway activation (i.e., C3a, C3b, C5a, C5b) are responsible for mediating many of the effects of the complement system (described in Complement effector molecules ).
Finally, although its role in human physiology and pathology has yet to be clearly demonstrated, complement activation can be triggered directly by proteolytic enzymes that cleave C3 and C5 to form C3a and C5a, respectively. This fourth extrinsic pathway can be initiated by enzymes of the coagulation cascade such as factors IX, X, XI/XIa, plasmin, and thrombin, as well as other enzymes, including cathepsin D, granzyme B, and β-tryptase, which are secreted by damaged cells or leukocytes. , Notably, there is also evidence for intracellular generation and function of complement activation fragments.
To maintain the balance between efficient destruction of pathogens and prevention of unwanted damage to host tissue, complement activation is tightly controlled by soluble and membrane-bound regulatory proteins. These include soluble factors such as carboxypeptidases; complement factors (CF)H, CFB, CFD, and CFI; C4b-binding protein (C4BP) and C1 inhibitor (C1inh); and membrane complement regulatory proteins (mCRPs) such as CD35 (complement receptor type-1; CR1), CD46 (membrane cofactor protein; MCP), and CD55 (decay-accelerating factor; DAF; Fig. 3.2 ). These CRPs protect both normal and neoplastic cells from damage by accelerating decay of convertases or cleaving activation fragments to inactive forms. Another membrane-bound protein, CD59 (protectin), binds to C8 and C9 to prevent assembly of the membrane attack complex (MAC). ,

Complement effector molecules
Activation of the complement cascade leads to generation of potent effector molecules, including C3a, C3b, C5a, and C5b.
Opsonin c3b
C3b not only amplifies the complement response via convertase formation (see Complement activation pathways ) but also acts as a mediator of innate and adaptive immunity. Cleavage of C3 induces a conformational change in the C3b fragment, allowing it to bind to proteins or carbohydrates present on cell membranes and foreign structures in a process called opsonization . C3b binding to CR1 on immune cells enables opsonized cells to be shuttled to the spleen and liver where C3b binds to the complement receptor of the immunoglobulin family (CRIg) expressed on tissue-resident macrophages such as Kupffer cells and induces phagocytosis. In addition to its role as an opsonin, C3b mediates adaptive immune functions, improving the contact between effector and target cells and potentiating antibody-dependent cell-mediated cytotoxicity (ADCC) and CDC. Additionally, C3b deposited on antigen-presenting cells interacts with CR1/CR2 expressed on antigen-specific T cells to promote their proliferation.
Anaphylatoxins C3a and C5a
C3a and C5a are powerful immune mediators through which the complement system exerts many of its effects. Small polypeptides comprising 77 and 74 amino acids, respectively, the anaphylatoxins have ∼36% overall homology but higher homology in the C-terminal “active” regions of the molecules. C5a binds two specific receptors, C5a receptor (R)1 (CD88) and C5aR2 (C5a-like receptor 2; C5L2), and C3a binds to a single receptor, C3aR. , All three receptors (C5aR1, C5aR2, and C3aR) belong to the superfamily of seven transmembrane spanning G protein–coupled receptors and are expressed primarily by myeloid cells, including monocytes, macrophages, eosinophils, basophils, and neutrophils. Expression by nonmyeloid cells has also been reported, especially in lung and liver. C5a binds both C5aR1 and C5aR2 with high affinity, but is thought to exert most of its biologic activity via the former.
C5a binding to C5aR1 downregulates cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling and activates signaling pathways such as phosphatidylinositol-3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) to induce a range of proinflammatory responses. These include chemoattraction of macrophages, neutrophils, basophils, and mast cells , ; enhanced phagocytosis ; and modulation of cytokine release. C5a triggers histamine release from basophils and mast cells, which in turn stimulates vasodilation and increased vascular permeability. It also stimulates neutrophil degranulation and release of toxic mediators such as reactive oxygen species (ROS) and neutrophil extracellular trap (NET) formation after priming with interferon (IFN)-γ. Additionally, C5a has been reported to stimulate angiogenesis by promoting the migration of microvascular endothelial cells. It also links to the adaptive immune system, influencing the trafficking and migration of B-cell populations , and modulating T-cell responses; it provides survival signals for naïve CD4 + cells, inhibits induction and function of regulatory T cells (Tregs), and promotes T-cell activation during interaction with antigen-presenting cells (APCs) in vitro and in vivo. The alternate receptor C5aR2 lacks G protein coupling and thus was originally thought to be a “decoy” or scavenger receptor, binding excess C5a without exerting direct physiologic effects. However, there is emerging evidence to suggest that C5aR2 can independently induce and moderate biologic functions of C5a through β-arrestin and p90RSK activation.
C3a has been reported to exert effects in mast cells, macrophages/monocytes, T cells, and APCs. It induces calcium mobilization from intracellular stores, , activation of extracellular signal-regulated kinases (ERK)1/2, and release of extracellular adenosine triphosphate (ATP) in monocytes and macrophages. Despite the lack of evidence for C3aR expression by T cells, C3a/C3aR has been reported to activate phosphoinositide-3-kinase (PI3K)-γ and induce phosphorylation of Akt, upregulating the antiapoptotic protein Bcl-2 and downregulating the proapoptotic molecule Fas, to decrease T cell apoptosis and enhance proliferation. However, the activity of C3a is short-lived, because it is rapidly cleaved at the C-terminal arginine to form C3a des-Arg, which can no longer bind to C3aR.
Compared with C5a, C3a is a much weaker chemoattractant but has been reported to exert a range of immunomodulatory functions including degranulation of eosinophils, basophils, and mast cells. , C3aR is thought to negatively regulate the mobilization of hematopoietic stem and progenitor cells from the bone marrow , and has also been shown to prevent neutrophil egress into the circulation, thus reducing acute tissue injury after ischemia or neurotrauma. Like C5a, C3a signaling may contribute to the regulation of adaptive immunity, inhibiting natural (n)Treg function and enhancing the survival and function of effector Th1 and Th17 cells. Conversely, the absence of C3aR signaling in CD4 + T cells is reported to be associated with enhanced interleukin (IL)-10, transforming growth factor (TGF)-β expression, and Foxp3 + -induced (i)Treg-mediated immunosuppression.
Both C3aR and C5aR have been shown to regulate Toll-like receptor (TLR)-induced cytokine production, with C5aR1 synergizing with TLR-2 and TLR-4 to elicit stronger inflammatory responses and C3aR regulating TLR9 signaling. , Moreover, TLR-induced inflammatory cytokines such as interleukin (IL)-6 can upregulate the expression of C3aR and C5aR.
Despite their critical roles in the development of effective immune responses, excess production of C5a and C3a can contribute to pathogenic proinflammatory responses, resulting in tissue damage and, eventually, multiorgan failure. Indeed, the anaphylatoxins are implicated in a range of inflammatory diseases including arthritis, ischemia-reperfusion injury, sepsis, neurodegenerative diseases, and cancer.
The membrane attack complex (MAC; C5b-9)
The membrane attack complex (MAC) is typically formed on the surface of pathogen membranes. Insertion of the MAC into the membrane of gram-negative bacteria, enveloped viruses, and parasites induces calcium ion influx and activation of lytic signals, which ultimately lead to cell death. Although insertion of the MAC into nucleated host cells can lead to membrane disruption and death by apoptosis or cell lysis, mammalian cells are typically resistant. This is because of either the shedding of MAC complexes deposited on the cell surface or the expression of regulatory factors, such as CD46, CD55, and CD59, which inhibit early complement activation and amplification and thus prevent MAC pore assembly.
In the absence of lysis, so-called sublytic MAC has been reported to signal through multiple pathways, including PI3K, Akt, and ERK, to exert different effects in different cell types. This includes effects on cell cycle and proliferation, apoptosis, protein synthesis, and membrane lipid composition (for in-depth review, see Morgan ). Sublytic MAC has also been reported to induce inflammatory cytokine production, trigger degranulation of neutrophils and macrophages, and induce platelet activation.
Complement therapeutics
Complement-targeting drugs have provided researchers with important tools to dissect the roles of the complement system in health and disease and also led to the development of powerful new therapeutic strategies (for in-depth review, see Mastellos et al., Ricklin et al., and Zelek et al. ). To date a number of complement therapeutics have been approved for clinical applications, mostly for rare (orphan) diseases such as paroxysmal nocturnal hemoglobinuria (PNH). These include drugs targeting serine proteases C1r, C1s, and MASPs (Cinryze, Cetor, Berinert, Ruconest), C3 (compstatin analogs such as pegcetacoplan), C5 (e.g., eculizumab), and C5aR1 (avacopan; Table 3.1 ).
| Target | Class | Agent(s) | Translational Status—Cancer | Translational Status—Other Conditions |
|---|---|---|---|---|
| C1r, C1s, MASPs | Serine protease inhibitor | C1-INH (Cinryze) | Not yet trialed | U.S. FDA and EU EMA approved for hereditary angioedema |
| C5 | Monoclonal antibodies | Eculizumab (Soliris), ravulizumab (Ultomiris) | Not yet trialed | U.S. FDA and EU EMA approved for PNH, aHUS, myasthenia gravis, neuromyelitis optica spectrum disorder |
| C5 | Oligonucleotide | Aptamer – ARC1905 | Not trialed | Trials discontinued |
| C5aR1 | Small-molecule antagonists | PMX53 (3D53, JPE1375), PMX205 | Preclinical | Phases Ia and Ib/IIa successful |
| C5aR1 | Small-molecule antagonists | CCX168 (avacopan) | Not yet trialed | U.S. FDA and EU EMA approved for antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis |
| C5aR1 | Monoclonal antibodies | IPH5401 (avdoralimab) | STELLAR-001, Phase I study of IPH5401, in combination with durvalumabin for advanced solid tumors | Phase II trial for severe COVID-19-related pneumonia unsuccessful |
| C5a | Monoclonal antibodies | IFX-1 (vilobelimab) | Not yet trialed | Sepsis, hidradenitis suppurativa, and COVID-19 Phase II trials completed |
| C5a and c5a desArg | Monoclonal antibodies | MEDI7814 | Not yet trialed | Preclinical |
| C3 and C3b | Compstatin analogs | Pegcetacoplan, AMY-101, APL2 | Not yet trialed | U.S. FDA and EU EMA approved for PNH |
| C3a | Competitive agonist | SB290157 | Preclinical | Preclinical |
Compstatin is a 13-residue cyclic peptide that selectively binds to human and primate forms of C3 and C3b, preventing propagation and amplification of complement activation and effector generation via all three pathways. Although the efficacy of the original drug was hampered by its limited in vivo half-life and the high plasma concentration of C3 (0.75–1.35 mg/mL), more potent and stable analogs have been developed, including AMY-101 (Amyndas Pharmaceuticals) and APL2 (Apellis Pharmaceuticals), which are in clinical trials for diseases such as periodontal, renal, neurologic, and ophthalmic diseases. , Indeed, APL2 (Pegcetacoplan or Empaveli; Apellis) was recently approved by the U.S. Food and Drug Administration (FDA) for the rare hematologic disorder PNH.
Eculizumab (Soliris) and ravulizumab (Ultomiris) are humanized monoclonal antibodies (mAbs) that target C5, blocking C5 cleavage to C5a and C5b and thereby preventing formation of the downstream MAC complex. The first complement therapeutic to be FDA-approved, eculizumab was approved for treatment of PNH in 2007, atypical hemolytic uremic syndrome (AHUS) in 2011, and more recently for generalized myasthenia gravis and neuromyelitis optica spectrum disorder.
Because they inhibit many of the main functions of the complement system (see Fig. 3.1 ), C3 and C5 inhibitory drugs are associated with an increased risk of infections. For example, approximately 50% of patients treated with eculizumab experience serious adverse events, including an increased risk of meningococcal disease. Additionally, because eculizumab is unable to block C5 cleavage mediated by the extrinsic protease pathway or in tissues that reside behind a restrictive barrier, a C5a-targeted approach may be preferable for specific conditions. The PMX family drugs are potent and highly selective C5aR1 antagonists developed at The University of Queensland, Australia. , These cyclic hexapeptides have properties favorable for clinical development, including small molecular weight (<1000 Da), nanomolar potency, plasma stability, oral bioavailability, and high receptor selectivity. The original drug, PMX53 (initially referred to as 3D53), was shown to effectively reduce C5a-mediated inflammatory responses in numerous animal models of disease, including ischemia-reperfusion injury, stroke, inflammatory bowel disease, atherosclerosis, arthritis, and epilepsy. Although Phase Ia and Ib/IIa safety/efficacy trials showed PMX53 to be safe, with no signs of toxicity, its poor oral bioavailability and short circulation half-life (around 20 minutes in humans) limited its clinical development. Medicinal chemistry improvements led to development of a lipophilic analog, PMX205, with enhanced efficacy and in vivo stability compared with its parent molecule. , A linear analog of PMX53 (JPE1375) has in vitro potencies comparable to those of PMX53, with improved receptor specificity, microsomal stability, and antagonistic potency in mouse but not human cells ; however, it also has a reduced in vivo half-life and pharmacodynamics. Other C5aR1 antagonists reported in the literature include CCX168 (avacopan), which is orally bioavailable, has a good safety profile, and was recently FDA-approved for the treatment of antineutrophil cytoplasmic antibody–associated vasculitis.
In addition to small-molecule drugs, monoclonal antibodies targeting C5aR1 have been shown to reverse inflammatory arthritis in a mouse model, and one of these antibodies, IPH5401 (avdoralimab), is being developed by Innate Pharma for the treatment of inflammatory diseases, including cancer. However, a Phase II clinical trial for treatment of severe coronavirus disease 2019 (COVID-19)-related pneumonia was suspended when it failed to meet primary endpoints. Antibodies targeting C5a are also in development, including IFX-1 (vilobelimab), which recently completed Phase II trials for treatment of sepsis, hidradenitis suppurativa (clinicaltrials.gov identifier NCT03001622), and COVID-19. Another antibody, MEDI7814, inhibits the binding of C5a and its less active desarginated metabolite (C5a des-Arg ) to both C5aR1 and C5aR2 receptors. Originally proposed as a potential therapeutic for acute inflammatory conditions, the antibody did not progress past Phase I clinical trial. However, it may be a useful tool to understand the interaction between C5a and the C5aR2 receptor. Finally, Mehta and co-workers used a mouse arthritis model to show that targeting C5 and C5aR1 simultaneously may be more effective than targeting either component separately.
Although a number of C3a agonists have been reported, the development of effective C3a inhibitors has proven more elusive. SB290157 is a nonpeptide arginine analog that acts as a competitive antagonist with high affinity for the C3a receptor. Antiinflammatory activity has also been demonstrated in animal models through a reduction in neutrophil numbers. However, like PMX53, SB290157 has a short circulation half-life. It has also been reported to have agonist properties and other off-target effects in a number of cellular systems, possibly depending on the level of receptor expression. New approaches to molecule development are underway and will potentially lead to efficacious and safe therapeutic agents. Of note, Lohman et al. reported a method (heterocyclic “hinge”) to convert small-molecule C3a agonists to antagonists by inducing conformational changes in the core structure. However, until the in vivo specificity, stability, and safety of these drugs can be confirmed, their therapeutic potential remains unknown.
Complement response to cancer
Bioinformatics analysis has identified the complement cascade as one of the key pathways associated with initiation and progression of cancers such as lung cancer, glioma, and hepatocellular and clear cell renal carcinoma. These findings are of great clinical interest, not only for identification of therapeutic targets but also for identification of potential biomarkers that could be used for screening, diagnosis, prognosis, and monitoring responses to therapy. The clinical potential of complement components as cancer biomarkers was highlighted by Lawal et al whose analysis of clinical data from 33 human cancer types showed that C3, C5, C3AR1, and C5AR1 expression is associated with tumor immune evasion via dysfunction or loss of T-cell phenotypes ; high C3 expression is also associated with shorter progression-free survival in several cancer types.
Evidence of complement activation in response to tumors comes from the detection of activation products including C3a, C4d, C5a, and C5b-9 in tumor tissue and plasma from human breast, , thyroid, lung, , oropharyngeal, esophageal, gastric, colorectal, and hepatic tumors. Complement proteins have also been detected in the ascitic fluid of ovarian carcinoma patients and C1q and C3 cleavage products deposited on the surface of tumor cells isolated from this fluid.
Tumor cells have been reported to activate all three pathways of the complement system, depending on the tumor type and the nature of the antigens expressed. Natural IgM leads to activation of the classical pathway through the recognition of tumor-specific antigens resulting from posttranslational modifications. For example, the expression of gangliosides GD3 and GD2 on the surface of melanoma and neuroblastoma cells leads to complement mediated cell lysis in vitro through this pathway. Local complement activation through the classical pathway has also been demonstrated in a murine lung cancer model. High mannose expression in glioma cell lines has been reported to activate the lectin pathway, and aberrant glycosylation patterns in murine sarcoma models activate classical or lectin pathways. Conversely, the alternative pathway may be activated by aberrantly expressed viral carbohydrates on the surface of virally transformed tumors such as Burkitt lymphoma or human immunodeficiency virus (HIV)-infected cells. The research thus far supports the core, but complex, role of complement in cancer progression and highlights the numerous therapeutic approaches available to modify tumor growth.
Complement dependent cytotoxicity and cancer
The complement system has traditionally been assumed to play a beneficial role in immune surveillance against tumors, contributing to antitumor defense mechanisms via CDC and ADCC. ADCC is mediated through the engagement of Fc receptors on natural killer (NK) cells and antibody mediated phagocytosis. CDC is initiated by binding of the Fc region of cell-bound antibodies to C1q, triggering classical pathway activation, MAC assembly, and direct cell lysis; at the same time, opsonic fragments (e.g., C3b, iC3b, and C3dg) deposited on tumor cells are recognized by complement receptors (CR1, CR3, CR4, or CRIg) on phagocytic cells, inducing phagocytosis and modulating the function of antigen-presenting cells. The release of inflammatory mediators such as C5a also contributes to the antitumor response by promoting recruitment of phagocytic cells to the tumor and upregulating the expression of Fc receptors on leukocytes to enhance antibody-dependent cellular cytotoxicity.
Unfortunately, naturally occurring antibodies to tumor-associated antigens are relatively weak and incapable of inducing efficient complement-mediated cytotoxicity. Moreover, vaccination attempts to induce production of high-affinity antibodies against tumor antigens have met with limited success in patients with cancer. However, the introduction of recombinant antibodies for cancer treatment has led to renewed interest in complement to promote antitumor defense ( Fig. 3.3 ). C5a agonists have been used as molecular adjuvants to induce antigen-specific antibody and cytotoxic T-lymphocyte (CTL) responses to weak antigens. For example, the C5a agonist (YSFKDMP(MeL)aR) was used in the murine B16 melanoma model to target C5aRs present on dendritic cells (DCs). This improved presentation of poorly immunogenic tumor-related antigens to T cells for subsequent cytotoxic T cell–mediated killing and reduced tumor growth. Another study demonstrated that the tumoricidal effect of a monoclonal antibody against human epidermal growth factor receptor 2 (HER2/neu) was augmented by fusion with C5a through enhanced recruitment of human granulocytes, the primary immune effector cells responsible for facilitating ADCC.
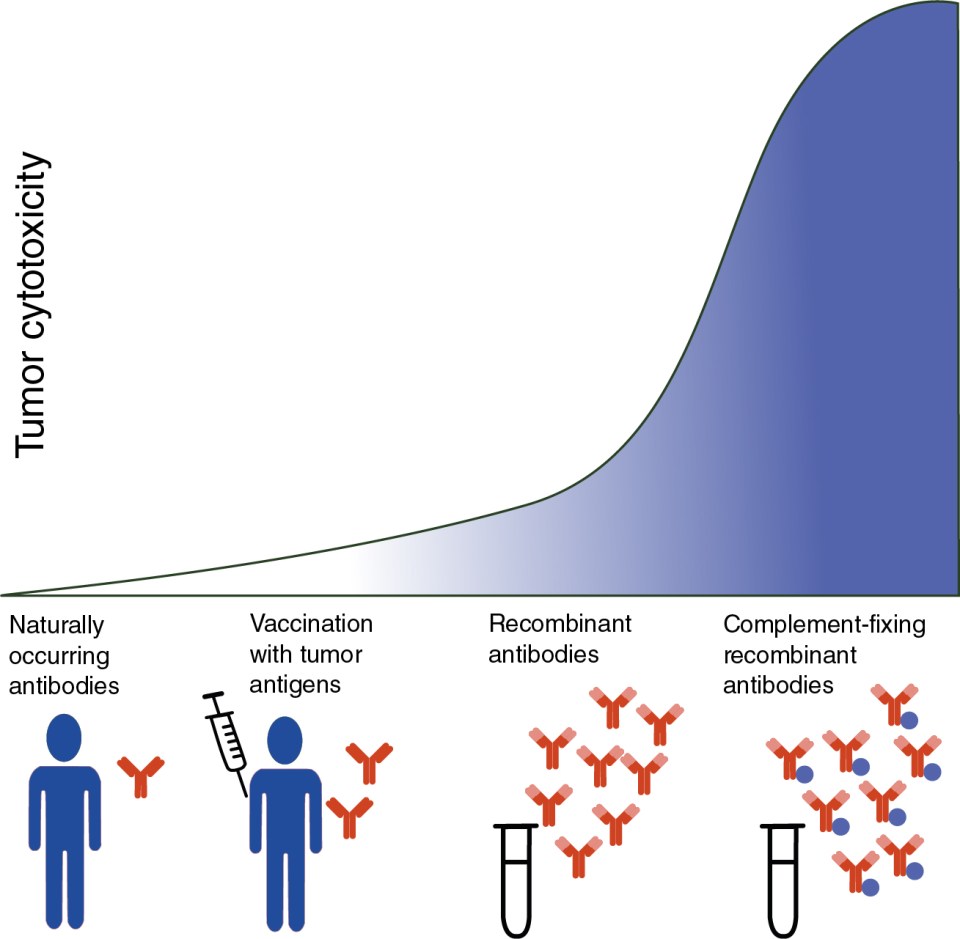
A successful strategy to optimize the therapeutic efficacy of mAb-based immunotherapies is the use of structural modifications to enhance complement activation and production of the MAC as an effector to kill tumor cells. , For example, the ability of anti-CD20 mAbs including rituximab to eliminate cancerous B cells in patients with chronic lymphocytic leukemia (CLL) through CDC is now well established. , Despite this, only a small number of the more than 35 mAbs approved by the FDA for anticancer treatment are complement-fixing, highlighting a possible window of opportunity to increase the efficiency of current immunotherapies (see Fig. 3.3 ). However, the efficacy of CDC for tumor targeting mAbs may be limited because of production of CRPs by cancer cells.
Complement regulatory proteins and cancer
Tumor cells are thought to escape immune attack by upregulating CRPs (for in-depth review, see Fishelson and Kirschfink ). These proteins act variously by inhibiting complement activation, blocking MAC assembly or insertion into the cell membrane, or facilitating removal of the MAC from the cell surface to protect against complement mediated lysis of tumor cells. High expression of membrane regulatory proteins such as CD46, CD55, , and CD59 , , on tumor cells is associated with increased metastatic potential and poor prognosis in many cancers (for in-depth review, see Geller and Yan ). Elevated expression of soluble regulators, CFH, and factor H-like protein (FHL)-1 in biologic fluids is also associated with poor outcomes in cancers such as ovarian, bladder, lung, and hepatic cancers. Other soluble regulators such as clusterin, factor I, and C4b-binding protein (C4BP) are secreted by tumor cells ( Table 3.2 ).
| Regulator | Function | Location | References |
|---|---|---|---|
| CD46 (MCP) | Cofactor for CFI–mediated cleavage of C3b and C4b | Membrane-bound | |
| CD55 (DAF) | Destabilizes C3/C5 convertases of the classical and alternative pathway | Membrane-bound | , |
| CD59 (protectin) | Inhibits the formation of MAC by binding C8 and C9 | Membrane-bound | , , |
| CFH | Decay accelerating activity and CFI cofactor in the alternative pathway | Plasma | , |
| Clusterin | Binds to C5b-7 and inhibits generation of C5b-9 | Plasma | |
| CFI | Cofactor dependent degradation of C3b and C4b. | Plasma | |
| C4BP | C3 convertase inhibitor in the classical and lectin pathway | Plasma |
CRPs may also limit the efficacy of therapeutic mAbs. For example, Golay and co-workers showed in freshly isolated patient’s cells that the efficacy of cell killing by the anti-CD20 mAb rituximab was dependent on the balance between levels of CD20 and CRPs such as CD55 and CD59. Accordingly, therapeutic strategies to block or silence CRP expression using monoclonal antibodies, RNA interference, or small peptides have been evaluated as a means of improving the therapeutic efficacy of antibodies such as rituximab. To this end, neutralization of CD55 and CD59 has been shown to enhance the antitumor efficacy of rituximab against leukemia and non-Hodgkin lymphoma cells. Neutralization of these same CRPs has also been reported to increase Herceptin-mediated complement cytotoxicity against lung cancer cells and eradicate micrometastases and small solid tumors from breast carcinoma and ovarian teratocarcinoma cell lines by complement-mediated mechanisms. Though this approach is limited by the ubiquitous expression of CRPs by normal cells as well as tumor cells, Macor and co-workers showed that bispecific Abs to CD20 and CD55 or CD59 specifically neutralized membrane CRPs on Burkitt lymphoma cells, enhancing their susceptibility to complement-mediated lysis in vitro and preventing tumor growth in a mouse model.
Complement activation in the tumor microenvironment enhances tumor growth
Although the complement system was originally thought to play an important role in cancer immunosurveillance, research over the past 15 years has revealed that complement activation within the tumor microenvironment suppresses effective antitumor immune responses and promotes tumor growth. Indirect evidence that complement activation products may contribute to tumor growth was provided in 2006 by Nozaki and co-workers, who showed that both C3aR- and C5aR1-deficient mice had reduced levels of neovascularization, vascular endothelial growth factor (VEGF) production, and blood vessel formation, all critical factors for tumor perfusion and invasion. However, the first direct evidence that complement proteins promote tumor growth came from a seminal study by Markiewski and co-workers in 2008 .
Role for C5a in regulating tumor growth
Markiewski et al. used a mouse cervical cancer model to show that C5a enhanced tumor growth via recruitment of myeloid-derived suppressor cells (MDSCs), leading to increased production of ROS and reactive nitrogen species (RNS) and suppression of CD8 + T cell–mediated responses. Since then, the tumor-promoting effects of C5a–C5aR signaling has been corroborated in numerous other murine cancer models, including lung, breast, ovarian cancer, lymphoma, and melanoma. The majority of studies suggest that C5a acts by promoting immunosuppressive myeloid cells, although the detailed mechanisms may differ between tumor types. For example, Corrales et al. demonstrated that C5aR1 antagonism with PMX53 inhibited lung cancer growth by reducing MDSCs and immunosuppressive molecules, including ARG1, CTLA4, IL-6, IL-10, LAG3, and PDL1 (genes for arginase 1, cytotoxic T-lymphocyte associated protein 4, interleukin-6, interleukin-10, lymphocyte-activation protein 3 and programmed death-ligand 1), within the tumor microenvironment. Although they showed that C5a induced endothelial cell chemotaxis and blood vessel formation in vitro, they found no differences in tumor vascular density in PMX53-treated mice. In contrast, Nunez-Cruz and co-workers found that C5a stimulates angiogenic activity by endothelial cells and that the absence of complement signaling impaired ovarian cancer growth, primarily by reducing tumor vascularization. Similar to previous reports, our laboratory showed that inhibition of C5aR1 signaling slowed the growth of B16 melanoma tumors by reducing tumor-infiltrating immunosuppressive leukocyte populations (MDSC, macrophages and Tregs) and increasing CD4 + T lymphocytes. We also provided evidence of a minor protective role for the alternate receptor, C5aR2, in modulating the effects of C5a on tumor growth, a result corroborated by Ding and co-workers, who showed that colorectal tumorigenesis was reduced in C5aR1-deficient mice but exacerbated in the absence of C5aR2.
C3a as a regulator of tumor growth
The first direct evidence that C3a contributes to tumor growth was provided by our laboratory, with the demonstration that growth of primary B16 melanoma was significantly reduced in C3aR-deficient mice. Daily treatment of established melanomas with the C3aR antagonist SB290157 also slowed tumor growth, suggesting the potential of C3aR as a therapeutic target. Investigations into the cells responsible for the antitumor responses showed that neutrophils and CD4 + T lymphocyte subpopulations were increased in the absence of C3aR signaling, whereas macrophages were reduced. The central role of neutrophils in the antitumor response was confirmed by antibody depletion experiments that reversed the tumor inhibitory effects observed in C3aR-deficient mice; tumor-infiltrating CD4 + T cells were returned to control levels, suggesting that neutrophils mobilized in response to C3aR inhibition may promote T-cell infiltration of the tumor.
Similar protective effects of C3aR deficiency/inhibition have been observed in other murine cancer models, including breast, colon, , lung, and sarcoma, although the cellular mechanisms may differ. An association between neutrophils and C3aR signaling in tumorigenesis was confirmed by Guglietta et al., who showed in a mouse model of spontaneous intestinal tumorigenesis that circulating lipopolysaccharide (LPS) induced complement activation and increased coagulation, neutrophil polarization, and neutrophil extracellular trap (NET ) formation. This group also demonstrated a correlation between neutrophilia and hypercoagulation in patients with cancer of the small intestine, suggesting the clinical potential of this research. The contribution of macrophages to the tumor-promoting effects of C3aR signaling has also been corroborated in mouse models of sarcoma and colon cancer, with C3aR deficiency shown to confer resistance to sarcoma and colon cancer growth via reduced macrophage accumulation, functional skewing toward M1-like phenotypes, upregulation of IFN-γ-associated genes, and enhanced T-cell responses. ,
As reported for C5aR, cellular responses to C3aR inhibition may vary, depending on the tumor type. For example, in orthotopic murine lung cancer models, C3aR inhibition was found to have no effect on myeloid cells, but increased IFN-γ + /tumor necrosis factor (TNF)-α + /IL-10 + CD4 + and CD8 + T cells. In contrast, the regression of murine mammary and colon tumors was attributed to increased cytotoxic NK cells. To gain further insight into the roles of cell populations within the tumor microenvironment, Davidson and co-workers used single-cell RNA sequencing to demonstrate that stromal cells play a key supporting role, producing C3 and promoting the recruitment and induction of immunosuppressive macrophages via C3aR signaling. Although they found no direct effects of C3a signaling on T cells, the authors suggested that disruption of signaling between stromal cells and infiltrating myeloid populations has the potential to affect subsequent interactions between the innate and adaptive compartments and thus improve the antitumor immune response.
Although the majority of studies have demonstrated protumor effects of C3a/C5a signaling, antitumor effects have been reported in some tumor types, most notably breast cancer models. For example, Kim and co-workers demonstrated that overexpression of C5a protected against the growth of EMT6 mammary tumors in mice. Tumor inhibitory effects of complement proteins were confirmed in an another (HER2/neu-driven) breast cancer model, in which C3 deficiency accelerated carcinogenesis. Further evidence that complement proteins protect against mammary tumor growth was provided by our laboratory; whereas C3aR/C5aR1 agonism slowed the growth of EMT6 and 4T1 mammary tumors, C5aR1 inhibition promoted their growth. , These results suggest that complement proteins can have different effects, depending on the cancer type, the site of the tumor, and the immune phenotype of the host. Compared with other common tumor models, 4T1 and EMT6 tumors have a low mutational load and are relatively immunogenic, with high levels of immune filtration. , The site of tumor cell injection may also influence the response because of differences in the nature of the tumor microenvironment and the ability of immune cells to infiltrate the site. Whereas 4T1 and EMT6 cells are injected orthotopically into the mouse mammary fat pad, many other commonly used tumor models are injected subcutaneously. The background immunophenotype of the host is also likely to be an important determinant of the response to immunotherapy. Whereas many tumor models are on a pro-inflammatory T helper lymphocyte (Th)1/M1 macrophage-oriented C57Bl/6 background, the HER2/neu transgenic model used by Bandini’s group, along with 4T1 and EMT6 mammary tumor models, is on an anti-inflammatory Th2/M2-oriented BALB/c background. Although C57Bl/6 and BALB/c mice have normal complement function, differences in complement activation levels within the tumor microenvironment are possible. As demonstrated by Gunn and co-workers in a mouse lymphoma model, low C5a levels promoted Th1 cell differentiation and reduced tumor burden, whereas high C5a levels promoted Treg differentiation and accelerated tumor progression. The complex role of complement proteins in cancer has been highlighted by Roumenina et al., who performed bioinformatics analysis of complement gene expression in 30 different cancers. This analysis showed that cancers could be classified into four groups, based on prognostic effect: (1) protective complement (favorable prognosis associated with high expression of complement genes), (2) protective C3 (favorable prognosis associated with high expression of C3 but not other genes), (3) aggressive complement (poor prognosis associated with high expression of complement genes), and (4) uncertain significance of complement (no obvious association with complement genes). Thus, understanding how different tumors and their microenvironments affect the response to complement-targeting (and other) immunotherapeutic strategies may provide critical insights relevant to future clinical application. This would include the identification of biomarkers to develop diagnostic tests that enable the stratification of patients for complement targeting therapies suited to their individual needs.
Sublytic MAC in cancer
The MAC (C5b-9) was originally thought to play an antitumor role through CDC. However, as described in Complement regulatory proteins and cancer , tumor cells have multiple strategies by which to protect themselves from MAC-mediated lysis. This includes the expression of high levels of CRPs CD46, CD55, and CD59, which act to limit complement activation and prevent assembly of the MAC. MAC can also be removed from nucleated cells by either budding off (ectocytosis) or engulfment (endocytosis). , These protective mechanisms may lead to low (sublytic) levels of MAC deposition that is insufficient to kill the target tumor cells but can affect cells in many ways. The assembly of sublytic levels of C5b-9 in the cell membrane increases cytosolic calcium and activates signal transduction pathways such as protein kinase C (PKC), PI3K-Akt, and ERK, increasing cell survival and proliferation and inhibiting apoptosis. It has also been reported to induce the release of inflammatory mediators such as ROS and RNS, leukotrienes, prostaglandins, inflammatory cytokines, adhesion molecules, growth factors, and matrix metalloproteinases (MMPs) and stimulate inflammasome activation by cells in vitro.
In the context of cancer, sublytic MAC has been shown to protect prostate cancer cell lines from TNF-mediated killing and induce the production of angiogenic growth factors by osteosarcoma cell lines. Vlaicu and co-workers have identified a downstream gene product, the response gene to complement-32 (RGC-32), as a potential regulator of tumor growth. RGC-32 contributes to cell cycle regulation by activating Akt and CDC2 kinases and is upregulated in many cancers, including colon and pancreatic cancers. , It has been shown to induce epithelial–mesenchymal transition (EMT) and to promote cancer cell migration and invasion in lung adenocarcinoma cells via reduction of matrix metalloproteinase activity. The limited in vivo studies suggest that RGC-32 is tumor suppressive in immunocompetent mice, but this effect may be reversed in tumors carrying TP53 mutations. Additional evidence that sublytic MAC contributes to tumor cell activation has been provided by Towner and co-workers, who used a bioinformatics approach to analyze the effects of sublytic MAC on the patterns of gene expression in MC38 colon cancer and B16 melanoma cells. The results revealed a downstream gene expression response likely to alter tumor behavior through induction of proliferative, migratory, and survival pathways, including a central role for EGFR signaling.
Roles for other complement components in cancer
Other complement proteins may also contribute to tumor growth. For example, deposition of C1q within the tumor microenvironment has been shown to accelerate tumor growth by promoting angiogenesis and regulating tumor cell motility and proliferation. In another study, C3 was shown to promote tumorigenesis independent of C3aR, C5aR1, C5aR2, C5, or terminal MAC, possibly through pathways mediated by iC3b/C3b on tumor-infiltrating myeloid cells. Indeed, iC3b has been shown to induce the expression of IL-10 and TGF-β2 and promote the generation of MDSCs from mouse bone marrow–derived dendritic cell cultures.
As described in Complement regulatory proteins and cancer , complement regulatory proteins can promote tumor growth by inhibiting complement activation and complement-mediated tumor cell killing. However, there is also evidence that negative complement regulators can protect against tumor development. For example, the negative complement regulator CFH has been shown to play a critical role in controlling spontaneous complement activation in the liver, with the absence of CFH leading to chronic inflammation and increased risk of hepatic carcinogenesis in mice; increased CFH expression is also associated with improved survival in patients with hepatocellular carcinoma. These results are in accord with a previous study investigating the role of pentraxin-related protein 3 (PTX3), a pattern recognition molecule that activates and regulates the complement cascade by interacting with C1q and CFH. PTX3 deficiency in mice was shown to increase susceptibility to mesenchymal (3-methylcholanthrene; MCA) and epithelial (7, 12-dimethylbenzanthracene; DMBA)/12-O-tetradecanoylphorbol-13-acetate; TPA) carcinogenesis via excessive complement activation, enhance C-C motif chemokine ligand 2/monocyte chemoattractant protein-1 (CCL2/MCP-1) production, and recruitment of tumor-promoting macrophages, leading Bonavita and co-workers to propose that PTX3 acts as an extrinsic oncosuppressor to limit tumor-promoting inflammation.
Autocrine effects of complement proteins
The majority of studies suggest that complement proteins influence tumor growth indirectly by modulating the immune response. However, tumor intrinsic effects are also possible. Evidence for a role for C5a–C5aR signaling in cancer cell invasion was provided by Nitta and co-workers, who demonstrated C5aR expression in human tumor tissue and human cancer cell lines, and that C5aR signaling enhanced cancer cell invasion in vitro by stimulating MMP release. A subsequent study by Cho et al. demonstrated expression of both C3aR and C5aR by ovarian cancer cells and that these receptors were capable of autocrine signaling via the PI3K/Akt pathway, leading to increased cell proliferation, migration, and invasion. This group also identified a role for Twist Family BHLH Transcription Factor (TWIST)1 in regulating C3 expression and suggested that C3a–C3aR signaling mediates EMT. Further support for a role for C5a in EMT came from Hu and co-workers, who showed that C5aR1 signaling downregulates epithelial markers E-cadherin and claudin-1 expression in hepatocellular carcinoma cells and upregulates the transcription factor Snail, a key inducer of EMT. C3a and C5a receptors have also been detected on human malignant hematopoietic cell lines and patient blasts; C3a and C5a stimulation of these cells enhances cell motility via activation of p38 MAPK and downregulation of heme oxygenase 1 (HO-1).
Autocrine signaling through the C3a receptor has also been implicated in the growth of some lung cancers and cutaneous squamous cell carcinomas, possibly via activation of the Wnt β-catenin pathway. Another study has suggested that reciprocal C3a-mediated paracrine signaling between cancer cells, cancer-associated fibroblasts (CAFs), and (possibly) myeloid cells promotes tumor progression and potentiates chemotherapy resistance. Though the majority of studies have focused on C3, C5, and their activation products, other complement proteins may also exert autocrine effects. For example, endogenous expression of C1r by cutaneous squamous cell carcinoma cells has been shown to promote tumor cell invasion through induction of MMP-13.
The role of complement activation in cancer progression
Tumor metastases are the primary cause of cancer deaths. In addition to effects on primary tumor growth, complement activation may contribute to metastasis at several stages: (a) modifying behavior of primary tumor cells via induction of EMT, leading to loss of cell–cell adhesion and increased motility , ; (b) enabling tumor cell invasion into blood or lymphatic vessels so they can enter the circulation and travel to distant sites; (c) increasing vascular permeability to facilitate tumor cell extravasation into organs such as lung, liver, and brain; and (d) contributing to the “premetastatic niche” in target organs by activating and recruiting immunosuppressive cells to facilitate seeding by the arriving tumor cells , ( Fig. 3.4 ).





