Monoclonal serotherapy
Robert C. Bast Jr. MD  Michael R. Zalutsky, PhD, MA
Michael R. Zalutsky, PhD, MA  Arthur E. Frankel, MD
Arthur E. Frankel, MD
Overview
Monoclonal antibodies have impacted significantly the care of patients with cancer. Some 22 monoclonal antibodies, cytotoxic drug conjugates, radionuclide conjugates, and targeted toxins have been approved by the US FDA for the treatment of a dozen different malignancies, although three of these biologicals are no longer marketed. Useful monoclonal antibodies have targeted proteins on the cancer cell surface most frequently (CD20 by rituximab and HER2 by trastuzumab and pertuzumab), inhibiting growth, inducing apoptosis, and enhancing chemotherapy, but some have targeted cytokines (IL-6 by siltuximab), growth factors [vascular endothelial growth factor (VEGF) by bevacizumab], and growth factor receptors (VEGFR2 by ramucirumab) that can affect endothelial cells in tumor vessels, or have altered the immune response by neutralizing checkpoint inhibitors (CTLA4 by ipilimumab and PD1 by nivolumab or pembrolizumab). In the case of trastuzumab and pertuzumab, binding of the two antibodies to different sites on HER2 cell surface receptors has produced greater antitumor activity than either alone. Enhanced cancer cell killing has also been achieved by conjugation of antibodies with cytotoxic drugs (emtansine to anti-HER2 trastuzumab and vedotin to anti-CD30 brentuximab) or radionuclide conjugates (90Y to anti-CD20 ibritumomab tiuxetan) permitting effective treatment for patients who had failed therapy with unconjugated antibodies. There are several barriers to effective therapy with monoclonal antibodies including antigen specificity, antigenic modulation, heterogeneity of antigen expression, effective delivery of antibodies to cancer cells, potency of effector mechanisms, and response to foreign globulin. The latter problem has been circumvented with the use of chimeric constructs, humanization of murine antibodies, and developing genetically engineered mice with the ability to develop fully human antibodies. Use of unconjugated antibodies is likely to improve as our knowledge of tumor biology and immunology grows, identifying targets such as OX-40 ligand. Use of smaller molecularly engineered binding constructs may improve pharmacokinetics and pharmacodynamics of serotherapy. Combinations of antibodies may be required to compensate for antigenic heterogeneity. Development of antibody drug conjugates will require the identification of monoclonal reagents that target tumor initiating stem cells. Pretargeting and the use of α-emitters are promising approaches to improving antibody-radionuclide conjugates. Development of targeted toxins must be further explored.
Introduction
Following the initial report of Kohler and Milstein,1 monoclonal antibody technology exerted a prompt and substantial impact on laboratory investigation. Over the past four decades, the availability of monoclonal reagents has permitted the development of novel markers for in vitro applications, including monitoring response to treatment, detecting malignant cells histochemically, identifying subsets of patients with particularly favorable or unfavorable prognoses, and distinguishing some tumors of unknown origin. Application of monoclonal antibodies for the in vivo treatment of human cancer has been more gradual, but serotherapy with monoclonal antibodies and their conjugates now has an established role in the management of a dozen hematopoietic and solid cancers.2–5
By 2015, the United States Food and Drug Administration (FDA) has approved 43 unconjugated monoclonal antibodies, drug conjugates, and radionuclide conjugates for therapeutic indications, including transplant rejection, coronary thrombosis, respiratory syncytial virus and anthrax infections, rheumatoid arthritis, systemic lupus erythematosus, paroxysmal nocturnal hemoglobinuria, macular degeneration, inflammatory bowel disease, psoriasis, asthma, and cancer.6, 7 Twenty-one of the 43 have been approved for the treatment of different cancers, as has one targeted toxin (Table 1). Three previously approved antibodies with anticancer activity have been withdrawn from the market. Unmodified monoclonal antibodies contribute to the care of patients with acute lymphoblastic and chronic lymphocytic leukemias (CLLs), Hodgkin and non-Hodgkin lymphomas (NHLs), Castleman disease, neuroblastoma, HER2-amplified breast cancer, non-small-cell lung cancer (NSCLC), head and neck cancer, renal cell cancer (RCC), colorectal cancer, and gastric cancer. With the general availability of these agents, it appears that monoclonal serotherapy has a well-established role in clinical oncology.
Table 1 Monoclonal antibodies, radionuclide conjugates, and targeted toxins approved in the United States for treatment of cancer7
| Antibody | Product name | FDA approved | Type | Antigenic target | Indication |
| Rituximab | Rituxan | 1997 | Chimeric | CD20 | Relapsed or refractory follicular and low-grade non-Hodgkin lymphoma |
| Trastuzumab | Herceptin | 1998 | Humanized | HER-2 | Metastatic breast cancers that overexpress HER-2 |
| Denileukindefitox | Ontak | 1999 | Humanized | CD25 | Cutaneous T-cell leukemia |
| Gemtuzumab | Mylotarg | 2000a | Humanized ADC | CD33 | Relapsed acute myelogenous leukemia in elderly patients |
| Alemtuzumab | Campath | 2001a | Humanized | CD52 | B-cell chronic lymphocytic leukemia |
| 90Y-ibritumomab tiuxetan | Zevalin | 2002 | Murine ARC | CD20 | Relapsed or refractory follicular and low-grade non-Hodgkin lymphomas in elderly patients and rituximab-resistant disease |
| 131I-tostuzumab | Bexxar | 2003a | Murine ARC | CD20 | Relapsed or refractory follicular and low-grade non-Hodgkin lymphomas in elderly patients and rituximab-resistant disease |
| Cetuximab | Erbitux | 2004 | Chimeric | EGFR | Metastatic colorectal cancer with irinotecan |
| 2006 | Head and neck cancers with radiotherapy | ||||
| Bevacizumab | Avastin | 2004 | Humanized | VEGF | Metastatic colorectal cancer |
| 2006 | Non-small cell lung cancer | ||||
| 2008 2009 2009 2014 | Advanced breast cancera Renal Cell Glioblastoma Ovarian |
aWithdrawn from the market.Source: http://www.antibodysociety.org/news/approved_mabs.php. Reproduced with permission of the Antibody Society.
In an attempt to exert greater antitumor activity in vivo, monoclonal antibodies have been linked to cytotoxic drugs, radionuclides, and immunotoxins. Extensive preclinical and clinical studies have now been performed with each type of immunoconjugate, with several approved by the FDA for the treatment of different cancers.8 This chapter considers the current use as well as some of the challenges and additional opportunities for the further clinical application of monoclonal reagents, drug conjugates, radionuclide conjugates, and targeted toxins for the treatment of patients with cancer.
Serotherapy for leukemia and lymphoma with unmodified monoclonal antibodies
With rare exceptions, murine monoclonal antibodies raised against human neoplasms recognize tumor-associated antigens, which are also expressed by normal adult or fetal tissues. Some antigens, however, are expressed by only a small number of normal cells that may not be essential to a patient’s well-being. Unconjugated monoclonal antibodies have contributed significantly to the care of patients with lymphoma3 and leukemia.9, 10
Anti-idiotypic antibodies
In the early 1980s, Levy and colleagues prepared tumor-specific murine monoclonal antibodies against the unique idiotypes associated with cell surface membrane immunoglobulin present on human B-cell lymphomas, but expressed by a very small subset of normal B cells.11, 12 Treatment of 18 lymphoma patients with anti-idiotypic antibodies alone produced an objective response rate of 67% with little toxicity, and one patient remained in complete remission for 72 months and survived for >17 years.6 In subsequent trials, anti-idiotypic antibodies were combined with interferon (IFN)-α, chlorambucil, or interleukin (IL)-2. Most of the antibodies that produced responses in vivo were of the murine immunoglobulin G1 (IgG1) isotype, which is generally least efficient in fixing complement or participating in antibody-dependent cell-mediated cytotoxicity (ADCC). Anti-idiotypic antibodies that bind to the B-cell receptor complex appear to induce apoptosis (programmed cell death) by delivering a death signal. In a fraction of patients treated with anti-idiotypic antibodies, recurrence of lymphoma is associated with loss of the relevant antigen from the cancer cells. Genes encoding the cell surface membrane immunoglobulin continue to undergo point mutations, resulting in the loss of idiotypic determinants.13 Use of anti-idiotypic antibodies has provided critical proof of concept, but widespread application has been hindered by the logistic challenge of developing reagents for each patient.
Anti-CD20 antibodies
Rituximab (Rituxan®)
Monoclonal antibodies against B cell differentiation antigens expressed by malignant cells from different patients have been used to treat NHL, as well as acute and chronic leukemias. One useful target is CD20, a 35-kDa phosphoprotein calcium channel expressed on the surface of all normal B cells and in 80% of NHLs but not on other normal tissues. Antibodies specific for different epitopes on CD-20 are classified as type I antibodies that translocate CD20 into detergent-insoluble lipid rafts in the cell membrane facilitating complement-dependent cytotoxicity (CDC) and type II antibodies that do not induce lipid rafts and facilitate ADCC by Fcγ receptor-bearing natural killer (NK) cells and macrophages.3 Repeated weekly administration of the type I chimeric mouse/human IgG1 anti-CD20 antibody rituximab produced a 48–50% response rate in patients with relapsed low-grade follicular NHL, with a median time to progression of 10.2–13.2 months.14, 15 In 37 newly diagnosed patients, treatment with rituximab produced an overall response rate (ORR) of 72%, with 36% complete responses and median time to disease progression of 2.2 years.16 Consequently, rituximab was used initially to treat patients with relapsed or refractory indolent follicular NHL. Use was extended to treat newly diagnosed low-grade NHL patients with rituximab alone or in combination with chemotherapy as well as for maintenance therapy.17 Rituximab has also contributed to the care of patients with diffuse large B cell NHL, CLL and autoimmune diseases.
Follicular and indolent NHL
Since regulatory approval by the US FDA in 1997 for the treatment of patients with recurrent or refractory follicular or indolent NHL, indications have been extended to provide 8 rather than 4 weekly courses and to retreat patients who had responded previously.18 After relapse, retreatment with a similar course of rituximab produced an overall response rate of 38% in 60 patients, with 10% complete remissions. Median time to progression exceeded 15 months.19 Combination of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy (R-CHOP) in 40 patients with low-grade follicular lymphoma, some of whom had been treated previously, resulted in an overall response of 100%, with 58% complete remissions and 42% partial remissions. Median time to progression exceeded 40.5 months.20 In a meta-analysis of 7 trials with 1943 patients with follicular lymphoma, other indolent lymphomas, and the more aggressive mantle cell lymphoma, the addition of rituximab to chemotherapy improved overall survival (OS), although the statistical significance was higher with indolent lymphomas than with the mantle cell histotype.21 The addition of rituximab to cyclophosphamide, vincristine, and prednisone (CVP) chemotherapy prolonged time to progression in patients with newly diagnosed follicular NHL, resulting in FDA approval in 2006 for use of rituximab in first-line therapy.22 Maintenance therapy with rituximab has prolonged progression-free survival (PFS) in four randomized trials, while OS has been extended in some but not all studies.23 In 2011, the US FDA approved rituximab for maintenance therapy based on a randomized comparison of rituximab (up to 12 8-week cycles) to no maintenance therapy in 1018 patients with high tumor burden follicular NHL who had complete or partial response to rituximab plus one of three chemotherapy regimens. Maintenance rituximab reduced the risk of a PFS event by 46% (P < 0.0001). At 3 years, PFS in the rituximab maintenance group was 74.9% versus 57.6% in the observation group (P < 0.0001).24
Diffuse large B-cell NHL
More aggressive diffuse large B-cell lymphoma (DLBCL) has been less responsive to rituximab alone. Among 54 patients with disease in relapse, the overall response rate to 8 cycles of rituximab was 31%, including 9% complete remissions with a median time to progression in responders of 8 or more months.25 Subsequently, 399 elderly patients with more aggressive DLBCL were randomized to receive R-CHOP or CHOP alone. A complete response rate of 76% was observed with the combination, compared with 60% with CHOP alone.26 Event-free survival (P < 0.005) and OS (P < 0.01) were prolonged significantly by the addition of rituximab. Similar results were obtained in two confirmatory trials,27, 28 both in young and elderly individuals, resulting in FDA approval in 2006 for use of R-CHOP in DLBCL, and providing the first improvement in the systemic treatment of DLBCL in two decades. In a recent meta-analysis of 7 trials involving 1470 patients with relapsed or refractory DLBCL, maintenance therapy with rituximab provided numerically improved PFS and OS, but the differences were not statistically significant, whereas incorporation of rituximab in salvage therapy led to statistically significantly better OS (P = 0.02) and PFS (P < 0.05) than rituximab-free regimens.29 However, the rate of infection-related adverse events was higher with rituximab treatment (RR = 1.37; P = 0.001).
CLL and Waldenstrom macroglobulinemia
Treatment with rituximab, alone or in combination with fludarabine, has been extended to CLL.30 In early studies, only a very modest response rate (15%) was observed with low standard doses of rituximab, possibly related to the lower concentration of CD20 on the CLL cell surface (8–15,000 vs 90,000 sites/cell) and to the shedding of soluble CD20, creating an “antigenic sink.”31, 32 Treatment with higher doses of rituximab or thrice weekly administration achieved an overall response rate of 46%, with an even higher response rate in previously untreated patients.31 A Cancer and Leukemia Group B (CALGB) trial of fludarabine and rituximab in 42 patients with CLL yielded a 100% response rate, with 48% of patients achieving complete remission.33 On the basis of the two randomized phase III studies where the addition of rituximab to fludarabine and cyclophosphamide extended PFS by 10–19 months in previously treated and untreated patients with CLL,34, 35 the US FDA approved rituximab in combination with fludarabine and cyclophosphamide in 2010. Rituximab has also been used with alkylating agents to treat Waldenstrom macroglobulinemia. One study reported a 74% response rate and 67% 2-year PFS with a combination of rituximab, dexamethasone, and cyclophosphamide.36
Autoimmune disease
Depletion of B lymphocytes has contributed to the management of autoimmune diseases. Rituximab has been approved in combination with methotrexate for the treatment of rheumatoid arthritis that has failed anti-tumor necrosis factor (TNF) therapy, as well as for treatment of granulomatosis polyangiitis (Wegener’s granulomatosis) and microscopic polyangiitis (MPA) with glucocorticoids.37
Toxicity
Rituximab therapy is generally well tolerated. Most side effects are infusion related and occur within the first few hours of treatment. Adverse events generally last minutes to hours and include chills, fever, nausea, vomiting, fatigue, headache, pruritus, and the sensation of throat swelling.38 Although side effects are experienced by up to 77% of patients, they are severe in only 10%.39 Normal B-lymphocyte counts can decrease to zero after initial infusion; recovery begins by 6 months and is generally complete by 9–12 months. As CD20 is not expressed on mature plasma cells, immunoglobulin levels are maintained, and intercurrent infections requiring hospitalization occurred in only 2% of patients during 1-year follow-up. Following FDA approval in 1997 and before 2002, >125,000 patients were treated with rituximab in the United States. Among these individuals, only eight deaths were associated with treatment related to the development of infusion reactions, paraneoplastic pemphigus, Stevens-Johnson syndrome, and toxic epidermal necrolysis.40 More than 120 cases of interstitial lung disease have been reported, generally when rituximab has been given with chemotherapy, but associated with monotherapy in 25%.41 In recent years, rituximab treatment has been linked to reactivation of hepatitis B virus (HBV) in HBsAg-negative/HBcAb-positive patients. In one meta-analysis of 578 patients across 15 studies, the risk of reactivation was estimated at 6.3%,42 suggesting that patients should be tested and antiviral prophylaxis given to patients with evidence of previous HBV infection who are receiving rituximab.
Mechanism of action and resistance
The mechanism(s) by which rituximab kills leukemia and lymphoma cells is not completely understood but probably involves ADCC, CDC, and the direct effect of CD20 ligation.43 In cancer cells, cross-linking CD20 can induce cell cycle arrest, inhibit DNA synthesis, activate caspases, and induce apoptosis.33 Sensitization to chemotherapy may relate to inhibiting the constitutive activation of AKT, thus downregulating the antiapoptotic protein Bcl-XL.44 NK cells, macrophages, and polymorphonuclear leukocytes can be important effectors for ADCC,33 and a correlation has been observed in some studies, between clinical response to rituximab and the presence of specific allelic polymorphisms in the FcγRIIIa and FcγRIIa receptors for IgG that are required to mediate ADCC.45 Individual NK cells are capable of “serial killing” of multiple lymphoma cells, particularly in the presence of rituximab that creates a “cap” at one pole of the cancer cell containing CD20, ICAM-1, and the microtubule-organizing center that enhances sensitivity to NK killing.3, 46 The response to rituximab is impaired in mice genetically deficient in C1q that lack the first component of the CDC pathway but that have intact ADCC.47 Clinical resistance to rituximab treatment rarely involves loss of CD20 expression but can be associated with upregulation of complement resistance proteins CD55 and CD59.48 Interestingly, different patterns of gene expression have been observed in lymphoma cells obtained before treatment from responders and nonresponders to rituximab.49 Gene expression in tumors that failed to respond resembled that in normal lymphoid tissues and exhibited higher expression of genes encoding certain complement components and genes involved in cytokine, T cell, and TNF signaling.
Ofatumumab (Arzerra®)
Ofatumumab (Arzerra®) is a type I human anti-CD20 IgG1 that binds to an epitope distinct from that recognized by rituximab with greater avidity and slower off-rates facilitating both CDC and ADCC. Ofatumumab was approved by the US FDA in 2009 for treatment of CLL based on a multicenter, randomized, open-label trial comparing ofatumumab in combination with chlorambucil to single agent chlorambucil.50 The trial enrolled 447 patients for whom fludarabine-based therapy was considered inappropriate by the investigator for reasons that included advanced age (median 69 years) or presence of comorbidities (72% with two or more comorbidities). Median PFS was 22.4 months for patients receiving ofatumumab in combination with chlorambucil compared to 13.1 months for patients receiving single-agent chlorambucil (P < 0.001).
Toxicity
The most common (>5%) adverse reactions with ofatumumab in combination with chlorambucil were infusion reactions, neutropenia, asthenia, headache, leukopenia, herpes simplex, lower respiratory tract infection, arthralgia, and upper abdominal pain. Overall, 67% of patients who received ofatumumab experienced one or more episodes of infusion reaction and 10% experienced a grade 3 or greater infusion reaction.
Obinutuzumab (Gazyva®)
Obinutuzumab (Gazyva®) is a type II humanized anti-CD20 IgG1 that acts predominantly through ADCC and has been glycoengineered to enhance binding to FcγR on effector cells.3 Binding of obinutuzumab to CD20 can initiate intracellular signaling, reorganizing actin fibers, increasing lysozomal membrane permeability, and producing nonapoptotic cell death.51 Obinituzumab was approved by the US FDA in 2013 for treatment of CLL based on a randomized open-label multicenter trial comparing chlorambucil alone and in combination with obinutuzumab in 781 previously untreated participants who were elderly or had comorbidities.52 Patients receiving obinutuzumab in combination with chlorambucil demonstrated a significant improvement in average PFS of 26.7 months compared to 11.1 months with chlorambucil alone.
Toxicity
The most common side effects in participants receiving obinutuzumab in combination with chlorambucil were infusion-related reactions, neutropenia, thrombocytopenia, anemia, musculoskeletal pain, and fever. Obinutuzumab was approved with a boxed warning regarding HBV reactivation observed with other anti-CD20 antibodies and rare cases of progressive multifocal leukoencephalopathy identified in participants on other trials of obinutuzumab.
Chlorambucil in combination with either obinutuzumab or ofatumumab provides an alternative for elderly CLL patients with comorbidities who are unlikely to tolerate fludarabine-based therapy.10
Antibodies against other lymphocyte-associated cell surface proteins
Over the past three decades, clinical trials have been conducted to evaluate antibodies against CD22, CD23, CD40, CD74, and CD80 in patients with different B cell-derived cancers.23 In early studies of CD10-positive acute lymphoblastic leukemia (ALL), anti-CD10 antibody-induced prompt modulation of the common ALL antigen, preventing effective therapy.53 Intravenous infusion of anti-CD5 also produced antigenic modulation and only transient, partial regression in a fraction of patients with T-cell leukemia/lymphoma and CLL.54 In one of the first studies of serotherapy with monoclonal reagents, a serum-blocking factor was demonstrated that prevented binding of the monoclonal antibody to circulating lymphosarcoma cells, consistent with the presence of shed tumor antigen.55
Antibodies against IL-6
Castleman disease is a lymphoproliferative disorder caused by release of IL-6 or other cytokines that can be localized to a single lymph node group or can be multicentric. Cytokine release can be stimulated by HHV-8 infection, but a fraction of Castleman disease cases are HHV-8 negative and the source of IL-6 has not been well defined.
Siltuximab (Sylvant®)
Siltuximab (Sylvant®) was approved by the US FDA and the EU in 2014 for treatment of HIV-negative, HHV-8-negative multicentric Castleman disease. Approval was based on an international, multicenter, randomized (2 : 1), phase 2 study comparing intravenous infusions of siltuximab and best supportive care (BSC) in 53 patients to placebo and BSC in 26 patients.56 Siltuximab produced a greater fraction of durable (18 weeks) tumor and symptomatic responses (34% vs 0%; P = 0.0012), tumor responses (38% vs 4%; P < 0.05), median time-to-treatment failure (>422 days vs 134 days; P < 0.05), and increased hemoglobin (36% vs 0%; p < 0.05) relative to the placebo controls. Despite success in the management of Castleman disease, siltuximab has failed to demonstrate clinical activity against other malignancies where IL-6 signaling is thought to be important, including multiple myeloma, RCC, and prostate cancer.57
Toxicity
Common adverse reactions (>10% compared to placebo) during treatment with siltuximab include pruritus, increased weight, rash, hyperuricemia, and upper respiratory tract infections. Consequently, siltuximab provides an excellent example of a targeted therapy against a cytokine that drives a rare, but debilitating lymphoproliferative disease.
Serotherapy for solid tumors with unmodified monoclonal antibodies
The HER family of transmembrane tyrosine kinase growth factor receptors has provided targets for serotherapy in solid tumors. As outlined in 108, interaction of peptide growth factor ligands with HER family receptors triggers signaling through the Ras-mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K) pathways, enhancing cell cycle progression, proliferation, and survival in normal cells and cancer cells. Of the four HER family receptors, most attention has been given to HER-1 (epidermal growth factor receptor or EGFR) and to HER-2.
Anti-EGFR antibodies
Several monoclonal antibodies have been prepared against the extracellular domain of the 170-kDa EGFR that is overexpressed in a number of carcinomas, including NSCLC, head and neck cancers, pancreatic cancer, and colorectal cancers.
Cetuximab (Erbitux®)
Cetuximab (Erbitux®) is a chimeric IgG1 monoclonal antibody that blocks the ligand-binding site of EGFR, preventing receptor activation, inducing internalization, and downregulating receptor levels. In experimental systems, treatment of human cancer cells with cetuximab produces cell-cycle arrest in G0–G1, induces p21, directs hypophosphorylation of Rb, inhibits proliferation, and blocks the production of angiogenic factors such as vascular endothelial growth factor (VEGF).58 In addition, treatment with cetuximab potentiates the activity of doxorubicin, paclitaxel, topotecan, and irinotecan as well as radiation therapy in nude mouse heterografts of human cancer. Potentiation of cytotoxic chemotherapy and radiation therapy may relate to inhibition of MAPK and PI3K with induction of BAX, activation of caspase 8, and downregulation of BCL-2 and NFκB, rendering cancer cells more sensitive to apoptotic stimuli.59 In addition, cetuximab can induce ADCC in the presence of peripheral blood mononuclear cells. Very little EFGR expression is required to mediate cancer cell death from ADCC.60 As in the case of rituximab, FcγR polymorphisms correlated with PFS after treatment with cetuximab, consistent with the importance of ADCC as a mechanism of cancer cell killing.61
Colorectal cancer
Weekly treatment with cetuximab alone produced partial remissions in 9% of 57 patients with chemotherapy-refractory colorectal cancer.62 In two larger trials, a combination of irinotecan and cetuximab was used to treat a total of 450 patients with documented metastases from EGFR-positive colorectal cancer who had received irinotecan previously.63 A combination of cetuximab and irinotecan produced a partial response in 17–23% compared with 11% of patients retreated with irinotecan alone. In one study, PFS, but not OS, was prolonged significantly from 1.1 to 4.1 months with the combination. In a subsequent phase III study, 1289 patients with recurrent EGFR expressing colorectal cancer who had been treated previously with first-line fluorouracil- and oxaliplatin-containing regimens were randomized to cetuximab plus irinotecan or irinotecan alone. Cetuximab improved disease-free survival significantly, but not OS, possibly related to cross-over in 47% of patients.64 Interestingly, the level of EGFR expression has not correlated with response to cetuximab-based therapy. Consistent with this observation, 4 of 16 previously treated patients (25%) with EGFR immunohistologically negative cancers responded to a combination of cetuximab and irinotecan.60 Consequently, patients should not be excluded from treatment based on immunohistochemical evaluation of EGFR. In 2004, the US FDA provided accelerated approval for cetuximab in combination with irinotecan to treat patients with irinotecan-resistant colorectal cancer. In 2007, regular approval was given, based on a trial where 572 patients with advanced EGFR-positive colorectal cancers that had failed oxaliplatin- and irinotecan-based regimens were randomized to BSC with or without cetuximab until progression. Cetuximab improved OS from 4.6 to 6.1 months (P = 0.0048).65 Approval was given also for use of cetuximab as a single agent to treat colorectal cancer that had failed oxaliplatin- and irinotecan-based regimens.
Similar to clinical results with small molecule inhibitors and other monoclonal antibodies reactive with EGFR, responses to cetuximab are rare in cancers with KRAS mutations.66 In retrospect, exclusion of patients with KRAS mutations affected the outcome substantiallyof the CRYSTAL trial where 1217 previously untreated patients with metastatic colorectal cancer were randomized to FOLFIRI alone or in combination with cetuximab, irrespective of KRAS mutation status.67, 68 Addition of cetuximab prolonged median PFS from 8.1 to 8.9 months (P = 0.036) and did not affect OS. When cancers were tested for KRAS mutations, the addition of cetuximab to FOLFIRI increased OS (19.5–23.5 months), PFS (8.1–9.5 months), and ORR (39% vs 57%) in patients with KRAS wild-type tumors. In patients with K-RAS mutant cancers, no improvement in OS, PFS, or ORR was associated with the addition of cetuximab to FOLFIRI. Supportive data were found in two other randomized trials where only patients with KRAS wild-type cancers benefitted,69, 70 resulting in approval in 2012 for cetuximab in combination with FOLFIRI for previously untreated patients with colorectal cancer.
Head and neck cancer
Cetuximab has also been approved for use in squamous cell carcinoma of the head and neck (SCCHN). In a pivotal multinational, phase III study, 424 patients with locally advanced head and neck cancers were randomized to high-dose radiotherapy or to a combination of radiotherapy with cetuximab.71 The addition of cetuximab increased the duration of locoregional control from 15 to 24 months and increased OS from 29 to 49 months. When cetuximab was administered to patients with recurrent SCCHN who had progressed on platinum-based therapy, response rates of 10–13% were observed over three prospective trials (n = 103) with disease control rates of 46–56%.72 The median time to disease progression ranged between 2.2 and 2.8 months, and the median OS ranged between 5.2 and 6.1 months.
Cetuximab has also improved the efficacy of platinum-based chemotherapy for SCCHN. Approval by the US FDA in 2011 was based on a multicenter trial in 442 patients with locoregionally recurrent or metastatic head and neck cancer who were randomly assigned to receive cisplatin (or carboplatin) with 5-FU with or without cetuximab.73 Significant improvements were seen in OS (10.1 months vs 7.4 months; P = 0.34), PFS (5.5 months vs 3.3 months; P < 0.0001), and objective response rates (35.6% vs 19.5%; P = 0.0001) in patients receiving cetuximab plus chemotherapy.
Toxicity
Side effects in a majority of patients included an acneiform rash, predominantly on the face and upper torso (Figure 1), and a composite syndrome of asthenia, fatigue, and malaise or lethargy. Treatment with minocycline can reduce the severity of the acneiform rash.74 Meta-analysis has found that the intensity of the rash correlates with OS, PFS, and ORR.75 Hypomagnesemia results from the direct effect of cetuximab on EGFR in distal renal tubules, producing magnesium wasting.76 A small minority of patients have experienced severe anaphylactic reactions, often on the initial infusion of cetuximab, related to a preexisting IgE antibody against galactose–α-1,3-galactose oligosaccharide found on the Fab portion of the cetuximab heavy chain.77
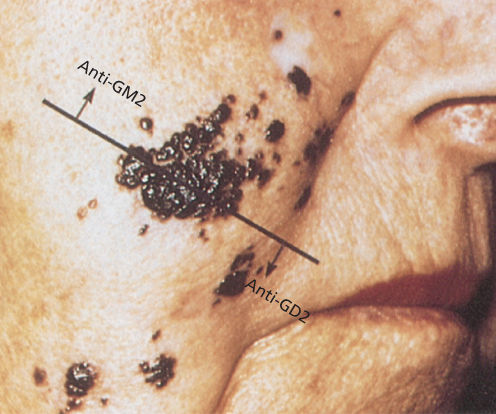
Figure 1 Recurrent melanoma, unresponsive to radiotherapy, before immunotherapy with intralesional injections of human monoclonal antibody to GM2 or GD2.
Panitumumab (Vectibix®)
Panitumumab (Vectibix®) is a fully humanized IgG2 anti-EGFR antibody that was given accelerated approval by the US FDA in 2006 based on a trial in 463 patients with EGFR-positive colorectal cancer resistant to standard drugs who were randomized to single-agent antibody therapy or BSC. Treatment with panitumumab produced an objective response of 10% compared to 0% in the BSC control group and extended mean PFS from 60 to 96 days.78 As with cetuximab, responses were limited to patients with wild-type nonmutated KRAS.79 Interestingly, in a recent meta-analysis, neither panitumumab nor cetuximab improved OS, PFS, or ORR in patients with HRAS wild type but BRAF mutant colorectal cancer, suggesting that sequencing BRAF might also identify patients who would not benefit from anti-EGFR treatment.80 Approval of the antibody by the EU for use in colorectal cancer occurred in 2007. In previously untreated recurrent colorectal cancer, attempts to add panitumumab to bevacizumab plus FOLFOX-4 or FOLFIRI were discontinued when interim analysis of >1000 patients showed a statistically significant advantage in the control arm without panitumumab.81
Toxicity
A similar spectrum of side effects has been observed with panitumumab and cetuximab. Acneiform rash and hypomagnesemia have been most notable.81 To date, fewer allergic reactions have been observed with panitumumab than with cetuximab.
Anti-HER-2 antibodies
Approximately 20–30% of breast cancers overexpress the 185-kDa tyrosine kinase growth factor receptor c-erbB2 (HER-2).82 While HER-2 lacks a functioning ligand-binding domain, it is the preferred dimerization partner for the other HER family members including EGFR, HER-3, and HER-4.83 Overexpression of HER-2 by breast cancer cells is associated with poor prognosis, particularly in node-positive disease, as well as with resistance to paclitaxel, CMF, and tamoxifen; however, it is associated with an improved response to doxorubicin.84, 85 Resistance to systemic therapy, increased risk of recurrence, and shortened survival reflect the biological consequences of HER-2 overexpression, including increased proliferation, increased cell survival, increased invasion and metastasis, and increased angiogenesis. Monoclonal antibodies directed against the extracellular domain of this receptor can inhibit growth of cancer cells that overexpress HER-2.86, 87 In addition, treatment with anti-HER-2 antibodies can increase the susceptibility of cancer cells to platinum compounds, taxanes, doxorubicin, and 4-hydroperoxy-cyclophosphamide.88, 89 Interestingly, binding of anti-HER-2 antibodies to HER-2 receptors in the juxtamembrane region90 can activate the tyrosine kinase87 but may prevent ligand-driven interaction of HER-2 with HER-3 to activate the PI3 kinase pathway, decreasing the antiapoptotic activity of phospho-AKT.87, 91, 92 Antibodies have been developed that bind to different sites on the HER-2 molecule.
Trastuzumab (Herceptin®)
Trastuzumab (Herceptin®) is a humanized IgG1 antibody that binds to subdomain 4 of the extracellular domain of the HER-2 receptor preventing homodimerization to other HER-2 receptors and downregulating HER-2 receptor levels. In vivo, inhibition of proangiogenic factors and mediation of ADCC may also play a role. Trastuzumab has received FDA approval for treatment of metastatic breast cancer, early breast cancer, gastric cancer, and gastro-esophageal junction (GEJ) cancer.
Breast cancer
In early clinical studies, trastuzumab produced objective regression of recurrent breast carcinoma in 12–15% of 269 heavily pretreated women.93, 94 Although cisplatin has demonstrated marginal activity against breast cancer in previous studies, a combination of cisplatin and trastuzumab produced an objective clinical response in 24% of 37 patients, with median duration of 8.4 months.95 A critical international multi-institutional study was performed in 469 women with recurrent breast cancer.96 Patients who had not previously received adjuvant therapy with doxorubicin were randomized to doxorubicin (or epirubicin) and cyclophosphamide, with or without trastuzumab. Women who had received adjuvant doxorubicin were randomized to paclitaxel with or without trastuzumab. The addition of trastuzumab to chemotherapy was associated with longer time to disease progression (median 7.4 months vs 4.6 months; P < 0.001), higher objective response rate (50% vs 32%; P < 0.001), longer duration of response (median 9.1 months vs 6.1 months; P < 0.001), and longer survival (median 25.1 months vs 20.3 months; P = 0.01). This study resulted in the approval of trastuzumab by the US FDA in 1998 and the EU in 2000 for the treatment of recurrent HER-2-overexpressing breast cancers. Subsequently, six large, multicenter adjuvant trials were undertaken (reviewed in Breast Cancer Neoplasms) to test whether the addition of trastuzumab improved the ability of chemotherapy to prevent recurrence of primary HER-2-positive breast cancer. Interim analysis in five of the six trials demonstrated sufficiently dramatic improvement in disease-free survival to terminate the clinical studies and to recommend use of trastuzumab in this setting.97–99 Addition of trastuzumab produced a 46–58% reduction in risk of recurrence, associated with an absolute reduction of 8–12% at 3 years in the 5 positive trials. Similarly, mortality was reduced 33–59%, producing an absolute decrease of 2–6% at 3 years.
On the basis of the results of two of these trials (NSABP B31 and NCCTGN9831) including 3752 women, the FDA granted approval in 2006 for the addition of trastuzumab to cyclophosphamide, doxorubicin, and paclitaxel for adjuvant therapy of women with HER-2-overexpressed cancer.
Trastuzumab enhances the response rate to several other cytotoxic agents used to treat breast cancer, including vinorelbine, gemcitabine, and platinum compounds.100–104 A randomized trial in 81 patients with metastatic HER-2-positive breast cancer who had not received chemotherapy for recurrent disease demonstrated a 51% response rate to vinorelbine and trastuzumab compared to a 40% response rate with paclitaxel and trastuzumab.105 In most studies, only those breast cancers with strong expression of HER-2, driven by gene amplification, responded to the antibody alone or to a combination of antibody with chemotherapy. Immunohistochemistry can provide an initial screen for HER-2 overexpression, but 2+ to 3+ reactions should be confirmed with the more reliable fluorescence in situ hybridization assay.106 HER-2 gene amplification can be acquired as breast cancers progress, arguing for repeated testing for HER-2 overexpression.107 Because only a fraction of patients responds, overexpression of HER-2 is necessary, but not sufficient reason to ensure response to trastuzumab. Lack of response to trastuzumab correlated with lack of expression of the PTEN phosphatase, the enzyme that removes phosphate groups from PI3 and interrupts signaling through AKT.108 Treatment with trastuzumab increased PTEN membrane localization and phosphatase activity by reducing PTEN tyrosine phosphorylation through inhibition of Src that could no longer dock on the HER-2 receptor.
Gastric and GEJ cancers
In 2010, the FDA granted approval for trastuzumab in combination with cisplatin and a fluoropyrimidine (either capecitabine or 5-fluorouracil) for the treatment of patients with HER-2-overexpressing metastatic gastric or GEJ carcinomas who had not received prior treatment for metastatic disease. The approval was based on results of a single international multicenter open-label randomized clinical trial, BO18255 (ToGA trial), which enrolled 594 patients with locally advanced or metastatic HER2-overexpressing adenocarcinoma of the stomach or GEJ.109 Patients were randomly assigned to receive trastuzumab plus chemotherapy or chemotherapy alone. The trial was closed after the second interim analysis, when the addition of trastuzumab was associated with improved median survival (13.5 months vs 11.0 months; P = 0.0038). An updated survival analysis demonstrated a persistent advantage to trastuzumab (13.1 vs 11.7) with the greatest benefit seen in HER-2 overexpressing cancers.
Toxicity
Treatment with trastuzumab is well tolerated and is associated with low-grade fever, chills, and fatigue that, generally, are observed with the first administration. In many studies, trastuzumab has been administered weekly, but it has been administered every 3 weeks at higher dosage with acceptable toxicity and trough levels.110 When trastuzumab has been combined with doxorubicin or paclitaxel, increased cardiotoxicity has been observed. In the pivotal trial that demonstrated the efficacy of trastuzumab in recurrent breast cancer, American Heart Association class III and IV cardiac dysfunction occurred in 27% of the group given trastuzumab with anthracycline and cyclophosphamide compared to 8% of the group given anthracycline and cyclophosphamide alone.111 A similar degree of cardiac dysfunction was observed in 13% of patients who received paclitaxel and trastuzumab compared with 1% who received paclitaxel alone. Long-term treatment of 218 breast cancer patients with trastuzumab-based therapy for at least 1 year was associated with an 11% incidence of class III cardiac dysfunction.112 In the six adjuvant trials where trastuzumab was given sequentially or concurrently with paclitaxel or carboplatin, but not doxorubicin, class III/IV cardiac dysfunction was observed in 0.5–4.1%.98, 99 Cardiac dysfunction generally responds to discontinuing trastuzumab and providing medical management. Thus, the benefits of trastuzumab for recurrent disease or adjuvant treatment generally outweigh the risks in patients with normal baseline cardiac function. The mechanism for trastuzumab-induced cardiac dysfunction remains obscure. Only low levels of HER-2 are found on cardiac myocytes, but trastuzumab can localize to the myocardium, and the ligand heregulin that binds to HER-2-HER-3 and HER-2-HER-4 dimers appears critical to the fetal development and survival of cardiac tissue under apoptotic stress.113 Use of less cardiotoxic anthracyclines in combination with trastuzumab offers one alternative. A neoadjuvant trial has been reported where concurrent epirubicin, paclitaxel, and trastuzumab produced a significantly higher pathological complete response (pCR) rate than did chemotherapy alone (67% vs 25%) without development of clinically evident congestive heart failure.114
Pertuzumab (Perjeta®)
Pertuzumab (Perjeta®) is an IgG1-humanized monoclonal antibody that binds to the dimerization domain of HER-2 at a site distinct from trastuzumab, preventing ligand-driven dimerization of HER-2 with multiple HER family members.115, 116 Use of pertuzumab in combination with trastuzumab synergistically inhibited survival of a breast cancer cell line that overexpressed HER-2 associated with increased apoptosis and blockade of signaling through AKT but not through MAPK.117 Pertuzumab was approved by the US FDA in 2012 and by the EU in 2013 for treatment of HER-2 amplified breast cancer based on a single clinical trial, Cleopatra, involving 808 patients with HER2-positive metastatic breast cancer who were randomly assigned to receive pertuzumab, trastuzumab and docetaxel, or trastuzumab and docetaxel with a placebo.118 Patients receiving pertuzumab had a median PFS of 18.7 months versus 12.4 months in the placebo group. With further follow-up, the addition of pertuzumab to trastuzumab and docetaxel increased OS by 15.7 months from 40.8 to 56.5 months (P < 0.001) and improved PFS by 6.3 months.119
In 2013, pertuzumab became the first FDA-approved drug for the neoadjuvant treatment of breast cancer in patients with HER-2-positive, locally advanced, inflammatory, or early-stage breast cancer (>2 cm in diameter or with positive lymph nodes) who are at high risk for recurrence and death. Pertuzumab was approved for use in combination with trastuzumab and other chemotherapy before surgery and, depending on the treatment regimen used, can be followed by chemotherapy after surgery. Following surgery, patients should continue to receive trastuzumab to complete 1 year of treatment. Pertuzumab’s accelerated approval for neoadjuvant treatment was based on a study designed to measure pCR in accordance with a new FDA advisory that this could be used as a surrogate endpoint. In the NeoSphere study, 417 participants were randomly assigned to receive one of four neoadjuvant treatment regimens: trastuzumab plus docetaxel, pertuzumab plus trastuzumab and docetaxel, pertuzumab plus trastuzumab, or pertuzumab plus docetaxel.120 About 39% of participants who received pertuzumab plus trastuzumab and docetaxel achieved pCR, compared to 21% who received trastuzumab plus docetaxel. The confirmatory trial for this accelerated approval is being conducted in participants with HER2-positive breast cancer who have had prior breast cancer surgery and are at high risk of having their cancer return. More than 4800 participants are enrolled in this trial, which will provide further data on efficacy, safety, and long-term outcomes. Results are expected in 2016.
Toxicity
The most common side effects reported in participants receiving pertuzumab plus trastuzumab and paclitaxel or docetaxel were hair loss, diarrhea, nausea, and neutropenia. Other significant side effects included decreased cardiac function, infusion-related reactions, hypersensitivity reactions, and anaphylaxis. There is a black box warning for cardiac failure and fetal damage.
Antiganglioside antibodies
GD2 is a disialoganglioside expressed on neuroblastomas and melanomas as well as on normal neurons, melanocytes, and pain fibers. Expression on cancer cells is relatively uniform and GD2 is not lost from the cell surface after treatment with monoclonal antibodies.121
Dinutuximab (Unituxin®)
Dinutuximab (Unituxin®) is a chimeric anti-GD2 IgG1 antibody that was approved by the US FDA in 2015 for treatment of newly diagnosed pediatric patients with high-risk neuroblastoma who have achieved at least a partial response to first-line multiagent, multimodality therapy. Addition of dinutuximab improved outcomes for patients who received maintenance therapy with granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, and 13-cis-retinoic acid (RA).122 The pivotal COG trial randomized 226 patients to either dinutuximab/RA or RA alone for six cycles of treatment. An improvement in event-free survival (EFS) (HR 0.57; P = 0.01, log-rank test) was demonstrated during follow-up and the trial terminated. At that time, the median EFS was not reached [3.4 years, NR, in the dinutuximab/RA arm, and 1.9 years (1.3, NR) in the RA arm]. An analysis of OS conducted 3 years later documented an improvement in OS in the dinutuximab/RA arm compared to the RA arm (HR 0.58), although median OS had not yet been reached in either arm.
Toxicity
The most common (>25%) adverse drug reactions in the dinutuximab/RA group were pain, pyrexia, thrombocytopenia, infusion reactions, hypotension, hyponatremia, increased alanine aminotransferase, anemia, vomiting, diarrhea, hypokalemia, capillary leak syndrome, neutropenia, urticaria, hypoalbuminemia, increased aspartate aminotransferase, and hypocalcemia. The most common (>5%) serious adverse reactions in the dinutuximab/RA group were infections, infusion reactions, hypokalemia, hypotension, pain, fever, and capillary leak syndrome.
Antivascular therapy
Angiogenesis is critical for normal fetal growth and wound healing but is also required for tumor growth and metastasis.123 Novel approaches to inhibiting angiogenesis have exploited the presence of antigens displayed on tumor-associated endothelium or the proangiogenic factors produced by tumor cells.
Bevacizumab (Avastin®)
Bevacizumab (Avastin®) is a humanized IgG1 that binds to the proangiogenic VEGF-A that has also been characterized as vascular permeability factor (VPF). Blockade of VEGF/VPF can inhibit tumor-driven angiogenesis in xenografts.124 Expression of VEGF/VPF has correlated with formation of ascites in mice with ovarian cancer xenograft models.125 Treatment with bevacizumab can completely inhibit ascites formation.126 In addition, cancer cells themselves can express VEGF receptors. Autocrine stimulation with VEGF can enhance proliferation and resistance to chemotherapy. Bevacizumab has received FDA approval for treatment of colorectal cancer (2004, 2006, and 2013), NSCLC (2006), breast cancer (2008; withdrawn 2011), RCC (2009), glioblastoma (2009), cervical cancer (2014), and ovarian cancer (2014), but its place in oncologic practice is still being defined.127
Colorectal cancer
In patients with previously untreated metastatic colorectal carcinoma, the addition of bevacizumab to irinotecan, fluorouracil, and leucovorin increased the overall response rate (34.8–44.8%) and significantly prolonged median PFS (7.4–10.4 months) and median OS (15.6–20.3 months).128 This trial led to the initial FDA approval in 2004 of bevacizumab for use with chemotherapy for first-line therapy of colorectal cancer. Two subsequent randomized phase 2 trials have demonstrated improved response rate, PFS, and OS when bevacizumab was added to 5-FU and leucovorin.129–131 In phase 3 studies where bevacizumab has been added to more effective first-line regimens, including FOLFOX-4 and XELOX, response rate, and OS were not significantly improved.132, 133 A number of combinations of bevacizumab and multiple drug combinations have provided similar results in noninferiority studies.134 In second-line therapy, however, addition of a higher dose of bevacizumab to FOLFOX-4 significantly increased response rate (9–23%), PFS (4.7–7.3 months), and OS (10.8–12.9 months) in bevacizumab-naive patients, leading to the approval of bevacizumab for second-line therapy in 2006.135 In patients with metastatic colorectal cancer who had progressed on bevacizumab in combination with irinotecan-based chemotherapy or oxyplatin-based regimens, continued use of bevacizumab in combination with the complementary chemotherapy regimen improved OS (11.2 months vs 9.8 months; P = 0.0062) and PFS (5.7 months vs 4.0 months; P < 0.0001), leading to approval for continued use of bevacizumab in second-line therapy.136
Non-small-cell lung cancer
In previously untreated metastatic NSCLC, three phase III trials have studied the addition of bevacizumab to carboplatin/paclitaxel,137, 138 or to gemcitabine/cisplatin.139 Significant increases have been observed in response rates (range 14–28.1%) with a more modest increase in PFS (0.6–2.7 months) or OS (0.5–2.0 months).134
Breast cancer
Addition of bevacizumab to paclitaxel in first-line treatment of patients with recurrent metastatic breast cancer significantly increased response rate (21–37%) and PFS (5.9–11.8 months).140 A modest, but significant, increase in PFS (8.0–8.8 months) was observed when bevacizumab was added to docetaxel.141 In second-line therapy, the addition of bevacizumab to capecitabine significantly increased the response rate (9–20%) but not PFS (4.2–4.9 months) or OS (14.5–15.1 months).142 Given the limited effect of bevacizumab in confirmatory trials, approval of the drug was withdrawn for breast cancer.
Renal cell cancers
A majority of sporadic RCCs exhibit inactivation of the von Hippel Lindau (VHL) gene with consequent overexpression of VEGF.143 In a randomized phase 2 trial that compared two doses of bevacizumab (3 or 10 mg/kg every 2 weeks) with placebo in previously treated patients with RCC, a significant prolongation of PFS was observed when high-dose bevacizumab was compared with placebo (2.5–4.8 months, P < 0.01).144 IFN-α is a standard initial therapy for RCC with a modest response rate and a survival advantage demonstrated in randomized trials. Two phase 3 trials have compared treatment with IFN-α and bevacizumab to IFN-α alone in previously untreated patients with RCC.145, 146 PFS was increased significantly from 5.2–5.4 to 8.5–10.2 months.
Glioblastoma multiforme (GBM)
Bevacizumab demonstrated activity against GBM (glioblastoma multiforme) in two single-armed trials, AVF3708g and NCI 06-C-0064E, where monotherapy produced a 20–25% response rate lasting a median of 3.9–4.2 months in a total of 141 patients who had relapsed after surgery, radiotherapy, and temozolomide.147 This prompted accelerated FDA approval for use in this setting. A recent double-blind, randomized trial evaluated whether the addition of bevacizumab to radiotherapy and temozolomide would improve outcomes in 637 patients with newly diagnosed GBM.148 Addition of bevacizumab increased PFS from 7.3 to 10.7 months (P = 0.007), but did not affect OS.
Ovarian cancer
In heavily pretreated patients with recurrent ovarian cancer, administration of bevacizumab, alone149, 150 or in combination with daily oral low-dose cyclophosphamide to provide “metronomic” therapy,151 has produced response rates of 16–24% with PFS of 4.4–7.2 months. Stabilization of disease for 6 months has been observed in approximately 40% of ovarian cancer patients. Four randomized studies have been performed evaluating the addition of bevacizumab to standard chemotherapy for front-line treatment (GOG 218152 and ICON7153) and for recurrent “platinum sensitive” (OCEANS154) and “platinum resistant” (AURELIA155) disease.156 PFS was improved in these studies by 3.8 months (P < 0.001), 1.7 months (P = 0.001), 3.3 months (P = 0.001), and 4.0 months (P < 0.0001), respectively. OS was not improved in any of the studies, but subsets of patients in GOG 218 and ICON7 with poor prognosis appeared to benefit. Lack of impact on OS overall and similar enhancement of PFS in first-line and recurrent disease has raised the question of whether treatment with bevacizumab should be delayed until disease recurrence.156 Approval for use of bevacizumab in combination with chemotherapy for recurrent disease was granted based on the Aurelia trial.
Cervical cancer
Addition of bevacizumab to a combination of carboplatin with paclitaxel or topotecan increased OS by 3.7 months (from 13.3 to 17.0 months, P = 0.004) and increased the rate of response from 36% to 88% (P = 0.008) in a randomized trial including 452 women with recurrent, persistent, or metastatic cervical cancer.157
Toxicity
In patients with NSCLC, RCC, colorectal, breast, cervical, and ovarian cancer, bevacizumab administration has been well tolerated by the majority. Grade 3 hypertension has been observed in approximately 20% of patients. While hypertension has been readily managed in most cases, malignant hypertension and fatal hemorrhagic stroke have been observed, arguing for aggressive monitoring and management of blood pressure. Significant proteinuria occurs in <5% of cases. Nasal bleeding has been observed. Greater risk for delayed wound healing and bleeding has been observed when bevacizumab was administered within 60 days of surgery.158 In patients with NSCLC, major hemoptysis was associated with 4 deaths among 35 patients in one early trial. Life-threatening hemoptysis occurred most frequently in elderly males with squamous cell histology, tumor necrosis, and cavitation, as well as disease close to major vessels. Patients with these characteristics have been excluded from many trials. Thromboembolic events have been observed in 5–7.4% of participants on randomized trials in ovarian cancer.156 Arterial thromboembolism has been observed in 2% of patients in large phase 3 trials across disease sites. In heavily pretreated patients with ovarian cancer, perforation of the bowel has been observed in 5–7% of cases, generally in the setting of partial small bowel obstruction and treatment response in lesions that involve the bowel wall. Bowel perforation has occurred in 2.6–3% of ovarian cancer patients on front-line adjuvant trials156 and in only 1% of colorectal cancer patients when bevacizumab was administered with FOLFOX.135
Ramucirumab (Cyramza®)
Ramucirumab (Cyramza®) is a human IgG1 monoclonal antibody that binds to the human VEGFR2 and prevents interaction with VEGF ligands. Ramucirumab has been approved by the US FDA for treatment of gastric and GEJ cancers (2014), lung cancer (2014), and colon cancer (2015).
Gastric and GEJ cancers
Ramucirumab can be used for treatment of fluoropyrimidine-resistant or platinum-resistant gastric or GEJ cancer as a single agent or with paclitaxel. Approval of ramucirumab as a single agent was based on a multinational, randomized double-blind trial in 655 patients with previously treated advanced or metastatic disease who were randomized (2 : 1) to ramucirumab or placebo plus BSC. Addition of ramucirumab to paclitaxel significantly improved OS (9.6 months vs 7.4 months, P = 0.017) and PFS (4.4 months vs 2.9 months, P < 0.001).159
Lung cancer
Ramucirumab in combination with docetaxel was approved for treatment of metastatic NSCLC that had progressed on platinum-containing regimens or anti-EGFR or anti-ALK targeted therapy. Approval of ramucirumab in combination with docetaxel was based on a double-blind, placebo-controlled clinical trial that enrolled 1253 patients with previously treated metastatic NSCLC.160 Addition of ramucirumab to docetaxel significantly increased OS (10.5 months vs 9.1 months; P = 0.024) and PFS (P < 0.001).
Colorectal cancer
Ramucirumab can be used in combination with FOLFIRI for the treatment of patients with metastatic colorectal cancer whose disease has progressed on a first-line bevacizumab-, oxaliplatin- and, fluoropyrimidine-containing regimen. This approval is based on the results of a randomized, double-blind, multinational trial enrolling 1072 patients who were randomly allocated to receive FOLFIRI plus placebo or FOLFIRI plus ramucirumab.161 Addition of ramucirumab to FOLFIRI improved OS (13.3 months vs 11.7 months; P = 0.023) and PFS (5.7 months vs 4.5 months; P < 0.001).
Toxicity
Ramucirumab treatment can be associated with fatigue, weakness, hypertension, hyponatremia, diarrhea, and nose bleeds. When combined with paclitaxel or docetaxel, neutropenia, febrile neutropenia, and anemia have been observed. Other rare, but important risks described in product labeling include hemorrhage, arterial thromboembolic events, infusion-related reactions, gastrointestinal obstruction, gastrointestinal perforation, impaired wound healing, clinical deterioration in patients with cirrhosis, and reversible posterior leukoencephalopathy. Hypothyroidism has been observed in patients with colorectal cancer.
Immune checkpoint inhibitors
Anti-CTLA4
Anti-CTLA4 monoclonal antibodies have been used to intervene in immunoregulation. CD4+CD25+ T regulatory (Treg) cells express cytotoxic T-lymphocyte antigen 4 (CTLA4). The presence of Treg cells in tumor tissue has been associated with a poor prognosis in several human cancers and their elimination can potentiate antitumor responses in preclinical models. In addition, effective activation of tumor immunity can be blocked by the interaction of CD80/86 on antigen-presenting cells with CTLA4 on T lymphocytes. Inhibiting this interaction with anti-CTLA4 antibody could enhance tumor-specific immunity.162
Ipilimumab (Yervoy®)
Ipilimumab (Yervoy®) is a fully human IgG1 monoclonal antibody that reacts with CTLA4 and was approved for use by the US FDA and the EU in 2011 based on a single international study of 676 patients with melanoma who had stopped responding to other FDA approved or commonly used treatments for melanoma and were randomized to ipilimumab, ipilimumab plus a gp100 vaccine, or vaccine alone. Those who received the combination of ipilimumab plus vaccine or ipilimumab alone lived an average of about 10 months, while those who received only the experimental vaccine lived an average of 6.5 months. Administration of ipilimumab as a single agent has produced a 7–15% objective response rate in human melanomas and RCC.163, 164 Greater activity might be anticipated using these reagents to augment the effects of specific tumor vaccines. Among 1861 patients, median OS was 11.4 months, which included 254 patients with at least 3 years of survival follow-up. The survival curve began to plateau around year 3. Three-year survival rates were 22%, 26%, and 20% for all patients, treatment-naïve patients, and previously treated patients, respectively.165 In an adjuvant study, 951 patients with completely resected stage III melanoma were randomized to ipilimumab or placebo. Median recurrence-free survival was prolonged by ipilimumab (26.1 months vs 17.1 months; P = 0.0013). Five (1%) patients died of drug-related events in the ipilimumab group.166 Response to CTLA4 blockade correlates with mutational load, neo-antigens, and expression of cytolytic markers.167, 168
Toxicity
Common side effects that can result from autoimmune reactions associated with ipilimumab use include fatigue, diarrhea, skin rash, uveitis, hypophysitis, endocrine deficiencies, and inflammation of the intestines (colitis) and hepatitis.164
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


