Aberrant signaling pathways in cancer
Luca Grumolato, PhD  Stuart A. Aaronson, MD
Stuart A. Aaronson, MD
Overview
Most, if not all, human cancers harbor aberrant activation of one or several signaling pathways, contributing to tumor initiation and/or progression. In this chapter, we describe how growth factors signal to receptors with intrinsic tyrosine kinase activity to promote cell proliferation and survival, largely through activation of the phosphatidylinositol-3′-kinase(PI-3-K)/Akt and Ras/MAP kinase pathways. Using different paradigmatic examples of oncogenic alterations in growth factor signaling, we discuss functional implications and relevance of these pathways for human cancer. General cell signaling principles in both normal and tumor cells will be addressed, and we briefly discuss how the initiation and progression of specific cancer types can be affected by the deregulation of certain other pathways, including those mediated by cytokines, the transforming growth factor-β (TGFβ) family, Wnt, Hedgehog, Notch, and nuclear receptors.
Finally, we discuss the concepts of oncogene addiction and targeted therapy and provide a few representative examples to illustrate how increased understanding of the mechanisms underlying signaling pathway aberrations in cancer can been translated to the clinic.
Intercellular communication in multicellular organisms is required for processes such as embryonic development, tissue differentiation, and systemic responses to wounds and infections. These complex signaling networks are in large part mediated by growth factors, cytokines, and hormones. Such factors can influence cell proliferation in positive or negative ways, as well as induce a series of differentiated responses in appropriate target cells. Cytoplasmic molecules that mediate these responses have been termed second messengers. The eventual transmission of biochemical signals to the nucleus leads to effects on the expression of genes involved in mitogenic and differentiation responses.
The pathogenic expression of critical genes in growth factor signaling pathways can also contribute to altered cell growth associated with malignancy. The v-sis oncogene of simian sarcoma virus, which encodes a growth factor homologous to the B chain of human platelet-derived growth factor (PDGF-B), is the paradigm for such genes.1 The normal counterparts of other retroviral oncogenes were subsequently shown to encode membrane-spanning growth factor receptors.2, 3 Other genes that act early in intracellular pathways of growth factor signal transduction have been implicated as oncogenes as well. Present knowledge indicates that the constitutive activation of growth factor signaling pathways through genetic alterations affecting these genes contributes to the development and progression of most, if not all, human cancers.
Because of space limitations, this chapter primarily focuses on growth factor signaling mediated by receptors with intrinsic tyrosine (Tyr) kinase activity. However, we briefly highlight other ligand-triggered pathways relevant to cancer biology.
Growth factor receptors with Tyr kinase activity
Hormones that act at great distances from the cells producing them have been known for many years. Hormones as signaling molecules were isolated from tissue fluids and readily characterized by their in vivo effects. The initial discoveries of growth factors indicated more subtle activities capable of stimulating the growth of chicken embryonic nerve cells in the case of nerve growth factor (NGF)4 or promotion of eyelid opening and incisor eruption in the case of epidermal growth factor (EGF).5 An important discovery concerning growth factors came from the demonstration of a unique enzymologic activity associated with binding of EGF to its receptor.5 Studies of the product of the viral oncogene, v-src, had led to the demonstration of its ability to act as a protein kinase.6, 7 Many protein kinases had been previously identified, but these had the capacity to phosphorylate serine (Ser) and/or threonine (Thr) residues. Moreover, it was well established that phosphorylations and dephosphorylations affected the activities of a variety of proteins. However, the src product was subsequently shown to have a unique specificity as a protein kinase in that it was capable of phosphorylating Tyr residues.8 Cohen then showed that addition of EGF led to phosphorylation of its purified receptor on Tyr residues.5 Subsequent studies have demonstrated that Tyr kinase activity is central to the functions of a large number of mitogenic signaling molecules.
More than 50 receptor Tyr kinases (RTKs) belonging to at least 18 different receptor families have been identified9, 10 (Figure 1). All RTKs contain a large, glycosylated, extracellular ligand-binding domain, a single transmembrane region, and a cytoplasmic portion with a conserved protein Tyr kinase domain. In addition to the catalytic domain, a juxtamembrane region and a carboxyl-terminal tail can be identified in the cytoplasmic portion. Because of their structure, RTKs can be visualized as membrane-associated allosteric enzymes with the ligand-binding and protein Tyr kinase domains separated by the plasma membrane. Their role is to catalyze the transfer of the γ-phosphate of adenosine triphosphate (ATP) to Tyr residues of exogenous substrates, as well as within their own polypeptide chain. Tyr phosphorylation represents the language that these receptors use to transduce the information carried by the growth factor.
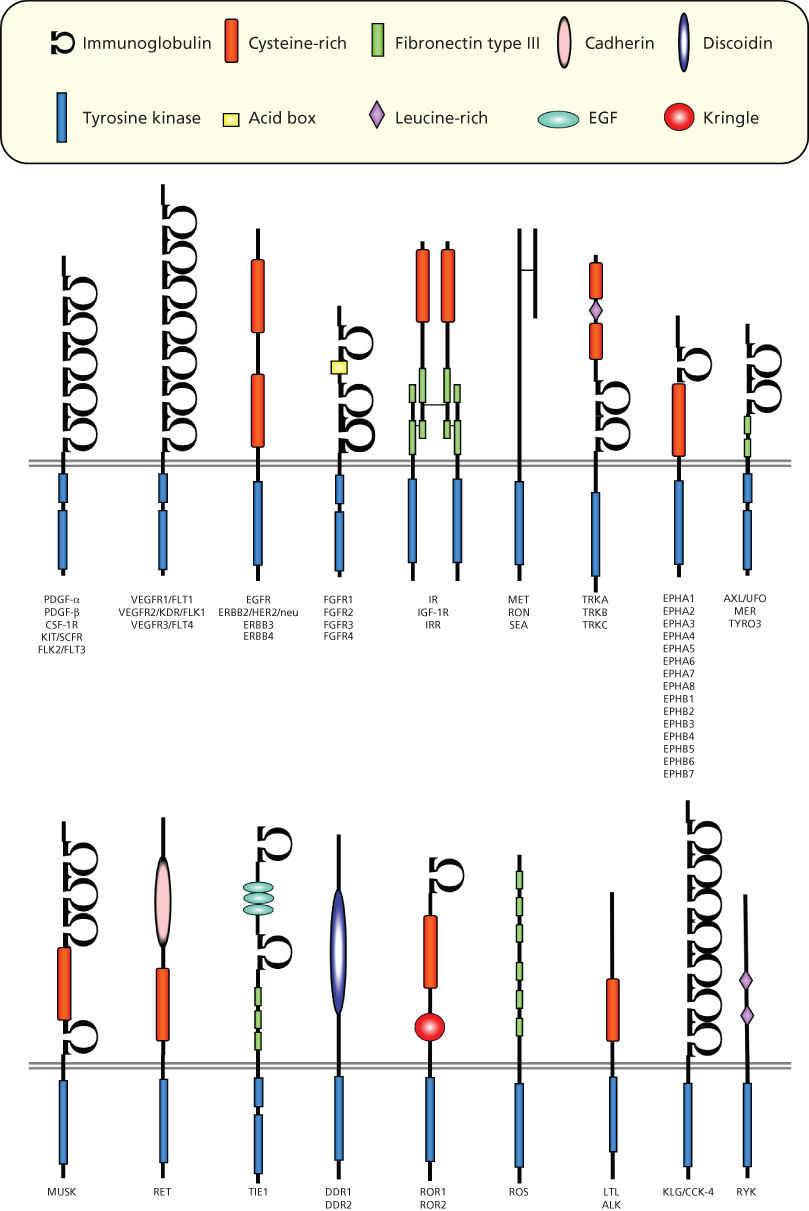
Figure 1 Families of receptor tyrosine kinases.
RTKs are activated by their ligands through receptor oligomerization, which stabilizes interactions between adjacent cytoplasmic domains and controls the activation of kinase activity.10 Dimerization can take place between two identical receptors (homodimerization), between different members of the same receptor family, or, in some cases, between a receptor and an accessory protein (heterodimerization),10 thus expanding both the repertoire of ligands recognized by each receptor and the diversity of effector pathways stimulated by a given receptor.
How ligands bind to induce receptor oligomerization varies for each class of RTKs.10, 11 PDGF, for example, induces receptor dimerization by virtue of its dimeric nature.12 EGF induces instead a conformational change in the extracellular domain of the epidermal growth factor receptor (EGFR), resulting in the exposure of a dimerization domain that is masked in the absence of the ligand.10 Fibroblast growth factor, which is a monomeric ligand like EGF, needs an accessory molecule, heparan sulfate proteoglycan, to induce receptor dimerization.13, 14 In contrast, the insulin receptor family exists as disulfide-bonded homo- or heterodimers of receptor subunits, and ligand binding causes a conformational change in the preformed dimeric receptor, which leads to receptor activation.15, 16
The activation of intrinsic protein kinase activity results in autophosphorylation of specific Tyr residues in the cytoplasmic portion of the RTK. Moreover, Tyr phosphorylation in the kinase domain stimulates the intrinsic catalytic activity of the receptor. Biochemical and structural studies have revealed some of the molecular mechanisms that mediate such activation. There is substantial evidence that autophosphorylation occurs in trans by a second RTK after dimerization induced by ligand binding. In the unphosphorylated state, the receptor possesses a low catalytic activity due to the particular conformation of a specific domain in the kinase region, which interferes with the phosphotransfer event. Phosphorylation of the kinase domain removes this inhibition, and the catalytic activity is enhanced and persists for some time independently of the presence of the ligand. Although kinase activity is at a low basal level in the monomeric state, this activity is sufficient to induce trans-autophosphorylation once the dimer forms. In addition to this mechanism, different RTKs contain cis-inhibitory elements outside the kinase domain, such as in the juxtamembrane domain (e.g., in KIT and MuSK) or the C-terminal tail (e.g., in Tie2), that can be disrupted by trans-phosphorylation following receptor dimerization. Besides the regulation of receptor catalytic activity, the other major function of RTK autophosphorylation is to create specific docking sites for downstream molecules that participate in the transduction of the signal (see the following section).
Aberrations affecting growth factor receptors in tumor cells
Although growth factor receptors can be constitutively activated by autocrine/paracrine loops, a number of other mechanisms have been identified by which growth factor receptors can become transforming. The paradigm for such alterations is the avian erythroblastosis viral oncogene v-erbB, which corresponds to a truncated form of EGFR, in which deletion of the ligand-binding domain results in the constitutive activation of the receptor.2 Alterations affecting a large number of RTKs have been implicated in different human malignancies. One mechanism involves the amplification or overexpression of a normal receptor, such as in the case of EGFR, ErbB-2, and MET.17–20 ErbB-2 was initially identified as an amplified gene in a primary human breast carcinoma and a salivary gland tumor.21, 22 Clinical studies have indicated that the normal ErbB-2 gene is frequently amplified and/or overexpressed in human breast carcinomas and in ovarian carcinomas,23 and detection in breast carcinomas of high ErbB-2 levels was initially a prognostic indicator of poor survival.17, 18 While ErbB-2 overexpression has been observed primarily in adenocarcinomas, the overexpression of a normal EGFR has been reported frequently in squamous cell carcinomas and glioblastomas.17, 24 In many cases, the EGFR appears to be activated by autocrine stimulation by one of its ligands, most commonly transforming growth factor-α.
Genomic alterations, such as mutation or rearrangement, have also been shown to activate the transforming capacity of RTKs in different human malignancies.25 For example, deletions within the external domain of the EGFR receptor or mutations in its Tyr kinase domain26, 27 are associated with its constitutive activation in some glioblastomas, lung and breast cancers.17, 24 The ret gene is activated by rearrangement, as a somatic event, in about one-third of papillary thyroid carcinomas, whereas germline mutations of cysteine residues in the extracellular region, which affect receptor dimerization, are responsible for multiple endocrine neoplasia (MEN) 2A and for the familial medullary thyroid carcinoma syndrome. Of note, point mutations in the RET kinase domain are associated with MEN 2B,28 indicating that the catalytic function of an RTK can be upregulated by oncogenic alterations affecting distinct domains of the receptor. Another example of an RTK activated by genetic alterations in the germline is the receptor for hepatocyte growth factor, MET, which is responsible for hereditary papillary renal carcinomas. Aberrant activation of MET through amplification/overexpression or mutations is also found in a variety of sporadic tumors including renal, liver, and gastric cancer.20 Finally, several other receptors, including the anaplastic lymphoma kinase (ALK), ROS1 and TrkA, have been shown to be oncogenically activated in human malignancies by chromosomal rearrangements that lead to fusion products containing the activated TK domain.25
Key Points
- RTKs are activated by growth factors through receptor dimerization and subsequent trans-phosphorylation.
- Tyr phosphorylation represents the language used by these receptors to transduce their signals, allowing specific recruitment of different effector or adaptor proteins.
- Different mechanisms are responsible for constitutive RTK activation in tumor cells. These include receptor/ligand overexpression, RTK amplification/mutation, and chromosomal translocation, which results in the formation of constitutively active fusion protein.
Signaling pathways of Tyr kinase receptors
Molecules, known as adaptor and scaffolding proteins,29 play an integral role in intracellular signaling by both recruiting various proteins to specific locations and assembling networks of proteins particular to RTK cascades. These adaptor proteins often contain a variety of motifs that mediate protein–protein interactions, including Src homology 2 (SH2) and phosphoTyr-binding domains, which bind to specific phosphorylated Tyr containing sequences, or SH3 domains, which recognize and bind to proline-rich sequences in target proteins.29, 30 One such adaptor, Grb2, is important in the activation of the small G-protein Ras (Figure 2).
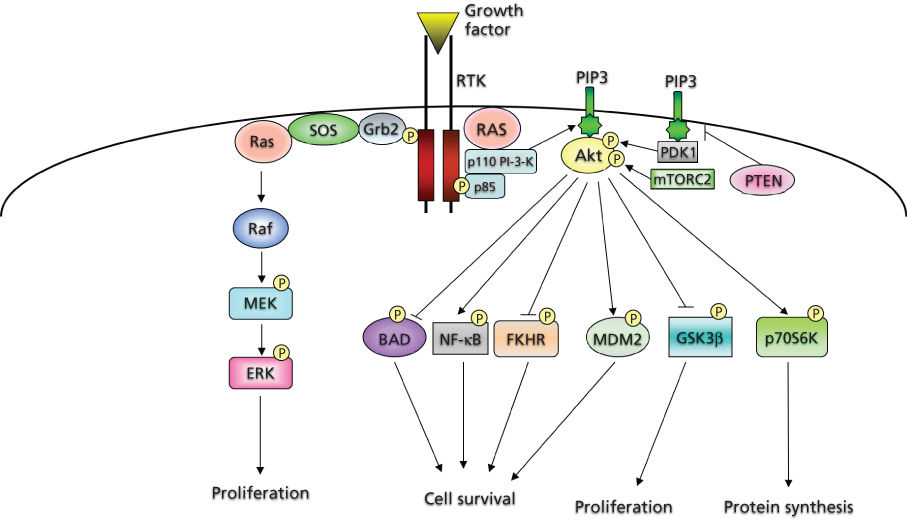
Figure 2 Intracellular effectors of receptor tyrosine kinases.
The PDGF system has served as the prototype for identification of the components of signaling cascades. Certain molecules become physically associated and/or phosphorylated by the activated PDGF receptor (PDGFR) kinase. These include phospholipase C-γ,31 PI-3-K regulatory subunit (p85),32 NCK,33 the phosphatase SHP-2,34 Grb2,35 CRK,36 RAS p21 GTPase-activating protein (GAP),37 SRC, and SRC-like Tyr kinases.38 Many of these molecules contain SH2 or SH3 domains.
PI-3-K and survival signaling
The regulation of cell survival and cell death is essential for both normal development and tissue homeostasis in adult organisms, where damaged cells must be removed and terminally differentiated cells must be sustained. A failure for this to occur may result either in the accumulation of mutations leading to cancer or to degenerative diseases.
PI-3-K is a lipid kinase that catalyzes the transfer of the γ-phosphate from ATP to the D3 position of the phosphoinositide (PtdIns), generating PtdIns(3,4,5)P3. These lipids can act in a variety of cascades, promoting the activation of several proteins (Figure 2). PI-3-K activation has been demonstrated to play an important role in survival signaling in a number of cell types.39, 40 The prototypical class 1 PI-3-K consists of two tightly associated subunits encoded by two distinct loci: a regulatory and a catalytic subunit. The classic mode of PI-3-K activation involves binding of the regulatory subunit to the phosphorylated Tyr residues of RTKs through SH2 domains, resulting in a conformational change that facilitates activation of the catalytic subunit. There are several known downstream effectors of PI-3-K. These include Rac, certain isoforms of protein kinase C, and, most relevant to cell survival, Akt, the cellular homolog of the viral oncogene v-Akt.39, 40 Three human homologs encode 57 kDa Ser/Thr kinases that contain an N-terminal pleckstrin homology (PH) domain, which binds to the activated PtdIns products of PI-3-K. These lipids are believed to mediate the localization of Akt to the plasma membrane. In addition, phosphorylation of two residues, Thr 308 and Ser 473, is required for its full activation.39, 40
Akt promotes survival and prevents apoptosis in various cell types through the regulation of different downstream pathways and effectors41 (Figure 2). Akt phosphorylates the pro-apoptotic protein BAD (BCL2-associated agonist of cell death), thus creating a docking site for the cytosolic protein 14-3-3, which sequesters BAD and inhibits its activity. Akt also phosphorylates FOXO transcription factors creating a binding site for 14-3-3, which retains them in the cytoplasm and inhibits their transcriptional targets, including pro-apoptotic proteins such as BIM and FAS ligand. Moreover, Akt can increase p53 degradation through phosphorylation of MDM2, and there is evidence that Akt can increase the pro-survival functions of the nuclear factor-κB (NF-κB) transcription factor complex and inhibit the pro-apoptotic c-Jun N-terminal kinase (JNK) and p38 pathways.41 Besides promoting cell survival, Akt can also exert positive effects on cell growth through inhibitory phosphorylations of the tuberous sclerosis complex 2, resulting in the activation of the mTOR pathway, or GSK3, which normally targets cyclin D1 for degradation41, 42 (Figure 2).
PI-3-K signaling in cancer
PIK3CA, which encodes the p110α catalytic subunit, was originally found to be amplified in a high percentage of ovarian tumors and ovarian tumor cell lines.43 Later studies revealed that PIK3CA is one of the most frequently mutated oncogenes in human cancer, particularly in colon, breast, endometrial, brain, and gastric tumors.44, 45 While far less frequently mutated in cancer, Akt can also play a role in human malignancies. Akt1 was found to be amplified 20-fold in a primary gastric adenocarcinoma.46 Additional studies have shown genomic amplification and overexpression of Akt2 in pancreatic and ovarian carcinoma cell lines, as well as in some ovarian and breast carcinomas.47 Overexpression of Akt2 occurs more frequently in undifferentiated and, thus, more aggressive tumors. Missense mutations in the Akt1 PH domain were identified in breast, colon, and ovarian cancers, resulting in prolonged Akt activation owing to its pathological localization to the plasma membrane.48
Further evidence of the involvement of the PI-3-K/Akt pathway in cancer stems from the discovery of the phosphatase and tensin homolog (PTEN) tumor suprressor, a gene inactivated by mutation in a high fraction of glial and endometrial tumors, as well as in melanoma, prostate, renal, and small-cell lung carcinomas.42, 45, 49 By dephosphorylating the three position of phosphatidylinositol, PTEN directly opposes PI-3-K activity, thus inhibiting Akt activation.
Ras
Ras proteins are a major point of convergence in RTK signaling and are important components of the cellular machinery necessary to transduce extracellular signals (Figure 2). These small GTP-binding proteins are membrane-bound intracellular signaling molecules that mediate a wide variety of cellular functions, including proliferation, differentiation, and survival. Ras acts as a molecular switch alternating from an inactive GDP-bound state to an active GTP-bound state. The paradigm for Ras activation involves the recruitment of a guanine nucleotide exchange factor (GEF) to the membrane in response to growth factor binding and subsequent activation of an RTK.50 GEFs promote the release of GDP from the catalytic pocket of Ras, and the relative abundance of intracellular GTP as compared to GDP ensures preferential binding of GTP. The best example of a Ras GEF is son of sevenless, which is brought to the membrane by its stable association with the adaptor protein Grb2.51 Although Ras is a GTPase, its intrinsic GTPase activity is actually quite low and requires additional proteins known as GAPs to promote GTP hydrolysis. GAPs can accelerate GTP hydrolysis by several orders of magnitude and are, thus, negative regulators of Ras functions.50, 52 An example of a Ras GAP is neurofibromin, encoded by the NF1 gene, which is frequently inactivated by mutation in different types of cancer, including glioblastoma and lung cancer, as well as in patients with the familial tumor syndrome, neurofibromatosis type I.50, 53
Ras mediates its multitude of biologic effects via a number of downstream effectors. Several proteins have been shown to directly bind to Ras in a GTP-dependent manner, including Raf, Ral, and PI-3-K.52 In fact, Ras can activate PI-3-K independently of RTKs, providing a direct connection between Ras and PI-3-K prosurvival signaling.50
Ras and cancer
Ras has been shown to be oncogenically activated by mutations in over 15% of all human tumors, and in some cancers, such as pancreatic carcinoma, the frequency is as high as 60%.52, 54 The initial evidence for Ras involvement in cancer came from the discovery of transforming retroviruses, Harvey and Kirsten sarcoma viruses, which contained H– and K-ras cellular-derived oncogenes. The first human oncogenes were identified by transfecting genomic DNA from human tumor cell lines into NIH3T3 mouse fibroblasts and isolating the DNA fragments from the transformed foci. These were shown to be the human homologs of the viral ras genes.55
The major hotspots for activating Ras mutations decrease the intrinsic rate of GTP hydrolysis by Ras and make the molecule significantly less sensitive to GAP-stimulated GTP hydrolysis. Thus, the outcome is a predominantly GTP-bound form that is constitutively active and essentially independent of growth factor stimulation.52, 54
Signaling downstream of Ras: Ras>Raf >MAPKinase cascade
The most well-studied effector of Ras is the Ser/Thr kinase Raf. Raf has been shown to bind to Ras and, in many cases, has been demonstrated to be indispensable for Ras functions, such as cellular transformation.56, 57 There are three known mammalian Raf isoforms, designated A-, B-, and C-Raf (also known as Raf-1), displaying distinct expression patterns.57 Ras-GTP binds to the amino terminus of Raf and promotes its activation, resulting in Raf homo- or heterodimerization and the phosphorylation of different Ser/Thr and Tyr residues participating in the regulation of Raf activity.57, 58 Once activated, Raf can phosphorylate the mitogen/extracellular-signal regulated kinase (MEK), also known as MAP kinase kinase (MKK), a dual specificity kinase, leading to its activation56, 58 (Figure 2). MEK can, in turn, activate MAP kinase or extracellular signal-regulated kinase (ERK) through tandem phosphorylations on both Thr 183 and Tyr 185. ERK then translocates to the nucleus, where it can activate a variety of proteins through phosphorylation on Ser or Thr, including the ETS family of transcription factors and the kinase p90 RSK, involved in protein translation.58, 59 Activation of the Ras-Raf-MAPK pathway results in an increase of DNA synthesis and cell proliferation, exemplified by the induction of cyclin D1,60 a major regulator of early cell cycle progression. In fact, cyclin D1 is also one of the most frequently amplified genes in cancer.61
Raf and cancer
The direct implication of Raf in human cancer came relatively recently with the identification of B-Raf mutations in more than half of melanoma cell lines and primary tumors.62 B-Raf mutations, which generally affect few specific residues, such as valine 600, were subsequently found in other malignancies, including thyroid (60%), colorectal (10%), and lung (6%) cancer.56 While B-Raf is one of the most commonly activated oncogenes, mutated in about 8% of all cancers, the frequency of A-Raf and C-Raf mutations is far lower, probably because of their weaker basal kinase activity compared to B-Raf.56 Of note, oncogenic activation of B-Raf can also occur following chromosome translocations, which result in the expression of fusion proteins containing the catalytic carboxy terminus of B-Raf. Such translocations have been found in pediatric astrocytoma, glioma, and thyroid cancers.56
Other MAP kinases
In addition to the ERKs, there are other MAP kinases belonging to distinct MAPK cascades with both different upstream activators and downstream effectors. JNK and p38 are stress-activated kinases involved in the cellular responses to a wide variety of extracellular stimuli, including mitogens, inflammatory cytokines, and UV irradiation.63 In contrast to its ability to activate the MAPK/ERK cascade, Ras only minimally perturbs JNK and p38, whose activity is mainly induced by the Rho family of small G-proteins, including Rac1, Cdc42, and RhoA. The pathways leading to JNK or p38 activation mirror those seen for ERK, with a variety of MKKs that can phosphorylate the various JNK isoforms.63
As with the ERKs, the end result of JNK and p38 activation is the phosphorylation of different transcription factors, resulting in increased expression of their target genes. Examples of transcription factors directly phosphorylated by either JNK or p38, or both, include ATF2, ELK1, JUN, MYC, and MEF2.63, 64 Of note, some of these factors were initially discovered as retroviral oncogenes.
Key Points
- Activated RTKs promote PI-3-K-mediated synthesis of PtdIns3P, resulting in the recruitment and activation of the kinase Akt. Through the regulation of different downstream effectors, Akt promotes survival in various cell types.
- RTKs stimulate cell proliferation largely through activation of the Ras/Raf/MAPK pathway.
- In human cancer, these downstream pathways can be frequently activated independently of RTKs, generally through mutations in the genes encoding various key effectors, including PI-3-K, Ras, and Raf.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


