Cancer cell immortality: targeting telomerase
Ilgen Mender PhD  Zeliha Gunnur Dikmen MD, PhD
Zeliha Gunnur Dikmen MD, PhD  Woodring E. Wright MD, PhD
Woodring E. Wright MD, PhD  Jerry W. Shay PhD
Jerry W. Shay PhD
Overview
Finding specific targeted agents for cancer therapy remains a challenge. One of the hallmarks of cancer is the limitless proliferation (immortalization) of cancer cells, which correlates with the activation of the ribonucleoprotein enzyme complex called telomerase. As 85–90% of primary human cancers express telomerase activity, while most normal cells do not, telomerase is a unique and almost universal therapeutic target for cancer. Although there have been several approaches developed for telomerase-targeted therapies, there is still no agent that has been approved. This chapter focuses on the pros and cons of the different approaches to target telomerase and summarizes preclinical and clinical results. We will also review novel telomerase-mediated telomere uncapping approaches that target telomerase-expressing cancer cells, but not normal cells.
Telomerase: a universal target for cancer therapy
Telomeres (TTAGGG)n are repetitive hexameric nucleotide repeats that are found at the ends of linear chromosomes. Telomere length varies in different organisms such as humans (2–15 kb) and mice (up to 100 kb).1 Telomeres are protected from being recognized as damaged or broken chromosomes by a special protein complex. This is termed the shelterin complex that stabilizes the chromosomal ends with 6 protein components (TRF1,TRF2, Rap1, TIN2, TPP1, and POT1) to protect the telomeric ends from exonuclease activity and DNA damage recognition.1–3 Shelterin proteins also have a role in the generation of a special lariat-like loop structure at each telomere end called the telomere loop (T-loop). The single-stranded telomeric DNA overhang at the ends of each chromosome bends backward and is believed to strand invade into the telomere DNA duplex structure disrupting the double-stranded TTAGGG repeats and forming a triple-stranded structure called the displacement loop (D-loop). The D-loop is bound by a single-stranded TTAGGG-binding protein called POT1 and this entire T-loop and D-loop structure functions in part by hiding or masking the single- and double-stranded telomeric DNA from being recognized as broken DNA and thus is important in telomere maintenance.4
G-quadruplex formation is a secondary structure that is generated between guanine bases in telomeric DNA and is also proposed to protect the ends of chromosomes from degradation.5 As normal somatic cells have a limited capacity to divide in culture, they stop proliferating after a finite number of cell doublings (the so-called Hayflick limit), which is related to DNA replication turnover.6 Because DNA polymerase is unable to replicate the 3′ end of the DNA lagging strand (often referred to as the end replication problem), in the absence of a maintenance mechanism, telomeres shorten with each cell division until they become “uncapped.”7 In addition to the end replication problem, oxidative stress and/or other unknown processes may accelerate telomeric shortening in vitro and in vivo. It is believed that only a few short telomeres with uncapped ends are sufficient to trigger a p53-dependent G1/S cell cycle arrest, which is known as replicative senescence. When cells enter senescence, the fate of normal cells is to remain in a quiescent phase. If other alterations occur in important cell cycle checkpoint genes such as TP53 or pRB, then senescence can be bypassed and cells continue to proliferate (extended lifespan phase) until many telomere ends become critically shortened leading to end fusions and chromosome bridge fusion breakage cycles (also referred to as crisis). Senescence and crisis are two fundamental mechanisms that protect cells against the early development of cancer. The cells that escape from crisis almost universally express the enzyme telomerase; a ribonucleoprotein cellular reverse transcriptase (Figure 1a), which has two essential components for the elongation of telomeres: the catalytic component hTERT (telomerase reverse transcriptase) and the telomerase RNA component (hTR or hTERC) (Figure 1b).8, 9 Telomerase is active during early human fetal development, then becomes silent in most tissues (Figure 2a) except embryonic stem cells and some but not all proliferating progenitors (e.g., male germline spermatocytes, activated lymphocytes, and a few other transient amplifying cell types).10 Telomerase is not absolutely required for cells to become malignant; however, 85–90% of primary human tumors have telomerase activity (Figure 2a), while the vast majority of normal tissues do not have detectable enzyme activity.9 A rarer (3–10%) mechanism to maintain telomere length is called ALT (alternative lengthening of telomeres) and involves intratelomeric recombination and perhaps T-loop resolution.
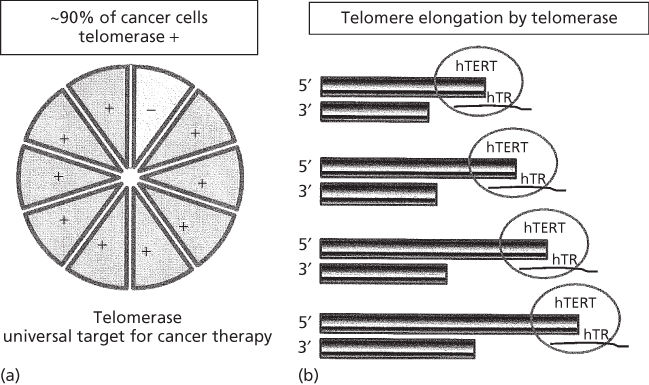
Figure 1 Normal cells that do not express telomerase activity cannot elongate their telomeres, hence their life span is limited whereas telomerase-expressing cancer cells (∼90% of cancer cells, a) can elongate their telomeres by telomerase (with hTERT and hTR subunits, b) resulting in unlimited proliferative lifespan (a hallmark of cancer). This important difference between normal and cancer cells make telomerase an almost universal therapeutic target.
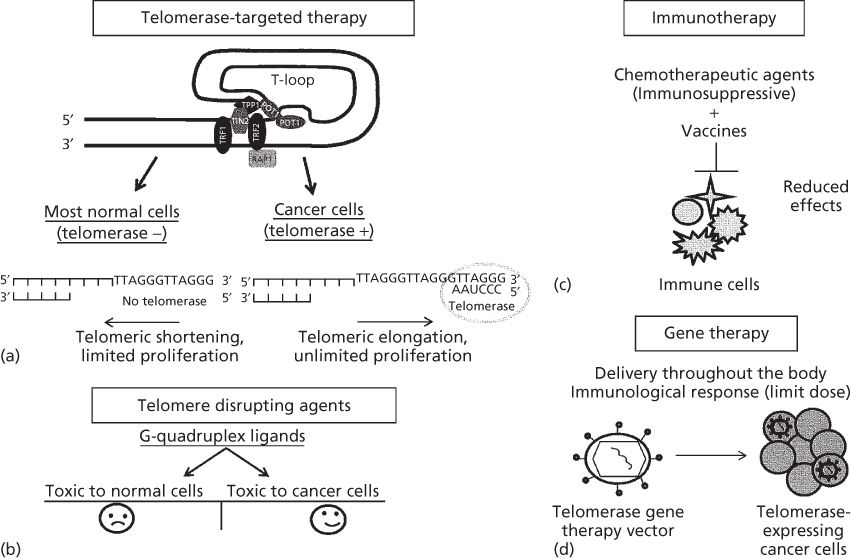
Figure 2 Diagram of hypothetical telomere demonstrating the t-loop structure. This structure is similar in normal and cancer cells but as normal cells generally do not express telomerase, telomeres shorten progressively while cancer cells expressing telomerase can maintain their telomeres (a). There are disadvantages of G-quadruplex stabilizers (b), immunotherapy (c), and gene therapy approaches (d). G-quadruplex stabilizers show their toxic effects by binding and stabilizing T-loop structures and they are also found at the other regions of chromosomes beside telomeres. Thus, these agents are likely to be toxic for both normal and cancer cells (b). One of the most important disadvantages for immunotherapy is that when combination chemotherapy is used with vaccines for cancer patients, some agents can block the effect of immunization as most chemotherapeutic agents are immunosuppressive (c). The main challenge for gene therapy is the delivery of the gene vector to the cancer cells (d) throughout the body. In addition, immunologic responses to therapy can also limit dosing. Gene therapies generally require more invasive approaches to delivery and are generally not orally available for systemic delivery.
There have been several studies on changes in telomerase expression/activation in normal and cancer cells. DNA rearrangement breakpoints within the TERT promoter region correlate with increased TERT expression in some cancers.11 In addition, mutations in the TERT promoter can lead to activated telomerase in immortalized, nontumorigenic fibroblasts.12 Different studies show that the mutations in the TERT promoter13 and gene amplifications in TERT14, 15 correlate with increased TERT transcriptional activation and expression of telomerase activity. While it is difficult to regulate very low abundant transcripts such as TERT, it has been reported that another mechanism for telomerase regulation is by alternative splicing. Precursor mRNA (pre-mRNA) that is transcribed from DNA is processed to exclude introns (noncoding sequences) and join exons (coding sequences) in order to produce mRNA for mature protein translation. However, during RNA splicing, exons may be included or excluded from the pre-mRNA to create diverse proteins with multifunctional roles. This process is known alternative splicing16 and may be important in the regulation of telomerase.
Induction of hTERT cDNA into most normal cells causes reactivation of telomerase and unlimited cell proliferation,17, 18 indicating that repression of telomerase activity is primarily regulated by transcriptional mechanisms and generation of full-length hTERT is likely responsible for activation of telomerase.19, 20 However, it is also known that posttranscriptional and epigenetic alterations may play an important role in this dynamic process.19 There remains no known consensus mechanism to repress telomerase during human developmental and to reactivate telomerase during cancer progression. Alternative splicing is one of the potential mechanisms for repression or reactivation of telomerase.21, 22 Several groups have found that there are different hTERT splice forms such as minus alpha, minus beta, or minus alpha beta,16, 21, 23, 24 which may have a role on regulation of telomerase activity.16, 20 hTERTα is one of the known splice variants that inhibits telomerase activity by acting as a dominant-negative inhibitor. Thus, the balance between hTERTα form and full-length hTERT and other splice variants may be important in the regulation of active telomerase.19, 25 Recent papers showed that a 1.1-kb region of a 38-bp variable number tandem repeats (conserved among Old World primates) is essential for exclusion of exon 7 and 8 to produce the minus beta splicing form and that RNA:RNA pairing between these repetitive sequences may be used to regulate decisions on hTERT splicing.26, 27 Importantly, misregulation of alternative splicing is a hallmark of nearly all cancers. In addition, a recent shift in our understanding of telomerase regulation has occurred. The common paradigm in the field was that hTERT was transcriptionally silenced in normal differentiated human tissues. However, there is increased evidence that hTERT is transcribed in normal cells but spliced to variants that do not have telomerase activity (Andrew Ludlow, unpublished observations). Although hTR (hTERC) is expressed in almost all cells, hTERT is only detected at extremely low levels even in cancer cells. How TERT is regulated to produce an optimal level for telomerase function in stem cells and cancer cells is still largely unknown. However, one could speculate that targeting hTERT at the splicing level may be a novel approach to reduce telomerase activity in cancer cells perhaps with minimal toxicity to normal cells.16
Compared to normal cells with long telomeres, the vast majority of cancer cells have relatively short telomeres. As normal stem cells generally have longer telomeres compared to cancer cells, this may facilitate cancer cells undergoing apoptosis with telomerase-targeted drug administration without a significant effect on normal cells.28 As an example, the transfection of an hTERT dominant-negative mutant into tumor cell lines resulted in apoptosis and reduced tumorigenic capacity.29, 30 The death by apoptosis in tumor cells correlates with initial telomere length, the tumor cells with short telomeres die before those with longer telomeres.31 In addition, it has been proposed that tumors are less likely to develop resistance against telomerase-based therapies compared to other cancer targets.32 However, this has not been experimentally observed in ALT or other telomere maintenance mechanisms. All of these factors suggest that targeting telomerase is not only a unique and almost universal cancer target, but it is likely to be a relatively safe anticancer treatment approach with fewer side effects compared to standard chemotherapy. Thus, a more complete understanding of the relationships between telomeres, telomerase, aging, and cancer may increase our knowledge and contribute to new approaches to cancer therapy and basic tumor biology research (Figure 1).
There are no telomere/telomerase-targeted drugs approved by the Food and Drug Administration (FDA)
Although proof of concept of different approaches against telomerase-based therapies has repeatedly been achieved in the preclinical setting, there are no telomerase inhibitors in clinical trials that have been approved by FDA. The most promising telomere/telomerase-targeted strategies are reviewed in Table 1.
Table 1 Telomere/telomerase-targeted strategies
| Therapeutic approach | Inhibitors |
| G-quadruplex stabilizers | BRACO19, RHPS4, Telomestatin, porphyrin, TMPyP4, CX-3543/quarfloxin, AS1411/ Cytarabine/Ara-C |
| Immunotherapy | GV1001, VX-001, GRNVAC1/2 |
| Gene therapy | Telomelysin, Ad-hTERT-NTR/CB1954, hTERTp-HRP/IAA |
| Telomere and telomerase-associated proteins | Geldanamycin (GA), Curcumin |
| T-oligo approach | PARP inhibitors |
| Small-molecule inhibitors | BIBR1532 |
| Oligonucleotides | GRN163L |
| Telomerase-mediated telomere uncapping approaches | Lentiviral mutant hTERC 6-thio-2′ deoxyguanosine |
G-quadruplex stabilizers
G-quadruplexes are formed by the ability of guanosine self-assembly together with monovalent cations (e.g., potassium) in the presence of guanine tandem repeats. G-quadruplexes may form in telomeres, oncogene promoter sequences, and other biologically relevant regions of the genome.33 It has been demonstrated that small molecules that target G-quadruplex structure in human telomeres cause telomerase inhibition (perhaps indirectly) and telomeric end disruption.34 Thus, this secondary structure is a popular target to design small-molecule ligands.
Telomestatin is isolated from Streptomyces anulatus 3533-SV4 and reported to be a telomerase inhibitor that interacts with G-quadruplex structures.35, 36 Nakajima and co-workers37 showed that telomestatin causes telomerase inhibition and induces apoptosis in primary leukemic blasts that were isolated from bone marrow of acute myeloid leukemia (AML-M2) patients. The other reported G-quadruplex ligand is termed TMPyP4. Hurley and colleagues showed that telomestatin caused telomerase inhibition by stabilizing of intramolecular G-quadruplex structures and eventually telomere shortening, whereas TMPyP4 suppressed the proliferation of ALT-positive cells as well as telomerase-positive cells by facilitating the formation of intermolecular G-quadruplexes leading to the induction of anaphase bridges. They also reported that telomestatin more tightly and specifically binds to the intramolecular G-quadruplexes compared to other G-quadruplex interactive compounds such as TMPyP4.38 Another investigated telomeric G4 ligand is termed RHPS4 (pentacyclic acridine). RHPS4 reduces telomerase activity at submicromolar levels through stabilization of G-quadruplex structures formed by single-stranded telomeric DNA.39 Others reported that RHSP4 causes telomere capping alterations in a telomere length-independent manner.40 In vivo experiments demonstrate that RHSP4 is even more effective when compared with other antineoplastic drugs in terms of antitumoral and antimetastatic effects.41 Although BRACO19 (trisubstituted acridine) is classified in the class of G-quadruplex stabilizers due to in vitro and in vivo anticancer effects,31 the utility of this compound is limited because of its low permeability for biological barriers in the airways or intestinal epithelial cell cultures in vitro.42 Several ligands induce growth inhibition of various cancer cells in vitro by causing cellular senescence and/or apoptosis. These ligands also exert short-term effects with telomere uncapping.43 Over the past decade, many G-quadruplex ligands have been described including fluorenons, bisubstituted acridines, cationic porphyrins, a perylenetetracarboxylic diimide derivative, indolo-quinolines. However, the evidence for specificity of these compounds for telomerase is very limited and they poorly bind to the quadruplex structures compared to double DNA.31 Thus, many of these compounds may affect quadruplex structures in normal cells as well as cancer cells (Figure 2b). Any compound that inhibits cell growth will decrease telomerase activity, so it is not sufficient to show telomerase inhibition to suggest specificity without also testing these compounds on normal cells without telomerase.
Some of the proteins that interact with G-quadruplex stabilizers also have the ability to induce senescence and/or apoptosis. Although G-quadruplex structures appear to be good targets for anticancer therapy, they are also observed during replication in chromosome regions besides telomeres. Because G-quadruplexes also have a large planar surface formed by terminal G-tetrad, most of the small molecules designed so far target this common structure instead of a specific target for a particular G-quadruplex. Thus, a general concern about these compounds is safety: while small-molecule ligands toward G-quadruplex structures affect cancer cells, they may also affect the telomere structure of normal cells44, 45 (Figure 2b).
Although there are two quadruplex-related small molecules that have already entered into clinical trials, there remains lack of direct evidence that these small molecules are specific for telomerase-targeted therapy. The first agent CX-3543 (quarfloxin/quarfloxacin, Cylene Pharmaceuticals, San Diego, CA, USA) is a fluoroquinolone derivative, that selectively disrupts nucleolin/rDNA G-quadruplex complexes in the nucleolus by inhibiting Pol1 transcription, resulting in decreased cancer cell proliferation and induction of apoptosis.46, 47 However, CX-3543 also interacts with MYC G-quadruplexes,46 so the in vitro antiproliferative and in vivo antitumoral activity in xenograft models may not be related to G quadruplexes at telomeres. In addition, CX-3543 selectively targets Pol1, rather than telomere function, which further suggests that CX-3543 may have indirect or even no effect on telomere function.47 The other quadruplex forming agent in clinical trials is AS1411 (Cytarabine/Ara-C, Antisoma Research, London, UK), which specifically targets nucleolin.48
Immunotherapy
Tumor-associated antigens (TAAs)-specific immunotherapy has been investigated for several decades. However, as most TAAs immunotherapies have been restricted to only a few tumor types, telomerase-based immunotherapy would theoretically be a good approach owing to the ubiquitous expression of telomerase in most tumor cells.49 Degradation of hTERT in proteasomes by E3 ubiquitin ligase generates protein fragments/peptides that may be presented on the tumor cell surface as antigens by the major histocompatibility complex (MHC) class I pathway. Thus, targeting these antigens by a patient’s own immune system (CD8+ T lymphocytes) is a direct way to kill the telomerase-expressing tumor cells50, 51 (reviewed in Ref. 52). Several hTERT vaccines have already been developed, but GV1001 and GRNVAC1 are the most promising vaccines that are in clinical trials (Table 2). GV1001 is a 16-mer TERT peptide (aa 611–626) that binds multiple HLA class II molecules. Recent studies showed that vaccination with hTERT-transfected dendritic cells helped to induce telomerase-specific CD4+ and CD8+ T-cell responses53 and also vaccination with GV1001-induced hTERT-specific T-cell responses in nonresectable pancreatic cancer patients and nonsmall lung cancer patients.54, 55 The combination treatment of GV1001 and cyclophosphamide in hepatocellular carcinoma failed to show partial or complete responses to treatment in a phase 2 clinical trial.56 GV1001 vaccination with chemotherapy together did not improve overall survival in advanced or metastatic pancreatic cancer in a phase 3 clinical trial.57 GRNVAC1 was also designed to induce CD4 and CD8 responses. GRNVAC1 was prepared from autologous dendritic cells transduced with mRNAs encoding a near full-length hTERT protein ex vivo. The dendritic cells are reintroduced into the patient and can potentially recognize any hTERT peptide in a patient’s tumor.32 Currently, GRNVAC1 is in phase 2 clinical trials for acute myelogenous leukemia patients. As monotherapy is likely to have limited efficacy, combination therapy might be a more effective approach to kill the heterogeneous nature of tumor cells. However, most chemotherapeutic agents are immunosuppressive and they might preclude the immunization effect. For this reason, vaccines should probably not be used in combination with chemotherapeutic agents58 (Figure 2c). Hence, new strategies are required to improve clinical efficacy with enhanced immune response effects of telomerase vaccination during chemotherapy.
Table 2 Telomerase-targeted vaccines in clinical trials
| Vaccines | ClinicalTrials.gov Identifier/phase | Condition | Drug intervention | Status |
| GV1001 | NCT01223209/phase 1 | Carcinoma | GV1001, LTX-315 | Completed |
| NCT01247623/phase1/2 | Advanced malignant melanoma | GV1001 | Completed | |
| NCT01342224/phase 1 | Locally advanced pancreatic adenocarcinoma | Tadalafil and vaccination | Active, not recruiting | |
| NCT00425360/phase 3 | Locally advanced or metastatic pancreatic cancer | Gemcitabine, capecitabine, GV1001 | Completed | |
| NCT01579188/phase 3 | Inoperable stage 3 nonsmall-cell lung cancer | GV1001 | Not yet recruiting | |
| NCT00509457/not provided | Nonsmall-cell lung cancer | GV1001 | Completed | |
| NCT00444782/phase 2 | Hepatocellular carcinoma | GV1001 | Completed | |
| NCT00358566/phase 3 | Advanced unresectable pancreatic cancer | GV1001, gemcitabine | Terminated | |
| GRNVAC1 | NCT00510133/phase 2 | Acute myelogenous leukemia | GRNVAC1 | Active, not recruiting |
| VX001 | NCT01935154/phase 2 | Nonsmall-cell lung cancer metastatic | VX001 | Recruiting |
Gene therapy
Gene therapy is a technique that treats diseases such as cancer by delivering therapeutic DNA into the patient’s cells. DNA that encodes a therapeutic effect can be delivered to the target cells. DNA becomes expressed by the cell machinery that ultimately makes a protein that can result in apoptosis. Genes can be delivered via oncolytic viral vectors and can include enzymes metabolizing prodrugs or suicide genes.59 Although gene therapy can be highly tumor specific, there are challenges including manufacture/formulation, which is often complex and clinical trials requiring special expertise60 (Figure 2d). Some studies showed the proof of concept in treating cancer, but there are some concerns about off-target effects. Targeting tumor cells in order to have a reduction of tumor burden and durable responses is the main purpose for successful therapy.61, 62 There are adenoviral vectors that have been engineered to kill cancer cells by only entering cells that have telomerase activity. These adenoviral vectors have a TERT promoter driving replication of the adenovirus. In this instance, the virus uniquely replicates in telomerase-positive cells, thus sparing most normal cells.63 After replication and amplification of the virus, the tumor cell will lyse and viral particles will spread to adjacent cells.63–66 This approach is known as a tumor-specific replication-competent adenoviral (hTERTp-TRAD) gene therapy approach.61, 67 One example for telomerase-mediated virus therapy is Telomelysin (OBP-301), which is an hTERT promoter driven modified oncolytic adenovirus. Preclinical studies demonstrated antitumor activity of telomelysin (and similarly engineered adenoviruses) in various cancer cells.64, 66, 68, 69 Phase 1 clinical trials did not show significant related toxic events but there were multiple manageable adverse events such as fever, chills, fatigue, and injection site pain. Some patients with solid tumors developed a decreased asymptomatic transient lymphocyte.70 Systemic treatment also has the potential effects on telomerase-positive proliferating normal stem cells. Therefore, systemic hTERTp-TRAD may affect transient amplifying stem cells such as proliferating cells in the crypts of the intestine, hematological cells, and a subset of cells in the basal and suprabasal layer of the epidermis.8, 60 The other concern is that intratumoral injections may not be sufficient to reduce the tumor burden.71
Suicide gene therapy (Ad-hTR-NTR), also called “gene directed enzyme prodrug therapy,” targets only tumor cells without affecting normal cells, similar to oncolytic gene therapy.32, 72 Three general steps are required for this approach: first is targeting the vector, second is transfecting the vector carrying an enzyme-encoding gene driven by hTERT into the cells, third is a prodrug that is activated by an enzyme, resulting in a release of toxins into the cytoplasm causing death of telomerase-positive cells.72 Although gene therapy (suicide gene or oncolytic vector) show some promising results, they do not appear to be as effective as vaccines or oligonucleotides.73 Future directions would be to make multifunctional viral vectors such as those replicating only in p53 mutant cells with telomerase expression. This would potentially reduce the effects on normal proliferating stem cells.
Small-molecule inhibitors and antisense oligonucleotides
Although millions of compounds have been screened during the past decade to identify small-molecule inhibitors of telomerase, there are currently no potent/specific small-molecule inhibitors except one candidate compound, BIBR1532. While BIBR1532 showed promising results in some preclinical models,74, 75 it was not effective enough to enter clinical trials perhaps owing to its short binding on/off rate with telomerase.76, 77 After many years of screening new drugs, GRN163L (Imetelstat, Geron Corporation) was found as a parallel approach to small-molecule inhibitors. This telomerase-targeted oligonucleotide approach uses a lipid-modified N3′ → P5′ thio-phosphoramidate oligonucleotide and has shown highly potent telomerase inhibition as a competitive enzyme inhibitor by being designed to complement the hTR template of telomerase that recognizes telomeres.78 GRN163L, 13-mer oligonucleotide (not antisense) was generated as a second improved generation of GRN163 to increase cellular uptake with a conjugating lipophilic palmitoyl (C16) group attached to the 5′ terminal.28 The effects of GRN163L have been investigated in different cancer cell lines and xenograft models. GRN163L demonstrates both in vivo and in vitro effects by inhibiting telomerase activity and resulting in progressive telomere shortening in a variety of cancer cell lines such as hepatocellular carcinoma,79 lung cancer,80 breast cancer,81, 82 bladder cancer,71 multiple myeloma,83 pancreatic cancer,84 colorectal cancer,85 glioblastoma,86 and esophageal adenocarcinoma.87 In vivo antimetastatic effects of GRN163L were tested in tumor xenograft models. When A549 lung cancer cells were injected into the nude mice via the tail vein, lung tumor formation was not observed in the animals treated with 15 mg/kg GRN163L (intraperitoneal, three times per week for 3 weeks).80 In mice having implanted hepatoma cancer cells, GRN163L inhibited tumor growth and also sensitized tumor cells to conventional chemotherapy.79 In a xenograft model of breast cancer and lung metastasis, administration of 30 mg/kg GRN163L for 12 treatments (three times per week, 4 weeks total) effectively reduced the proliferation of breast cancer cells and metastasis to the lungs.82 Different studies showed that GRN163L reduced tumor size as well as improved survival in a human myeloma cell-derived xenograft model.83 Although there are many studies showing promising effects of GRN163L in vitro and in vivo, GRN163L has not succeeded in most clinical trials owing to hematological side effects and liver function abnormalities as well as other adverse events. When patients had dose-limiting toxicities such as thrombocytopenia, owing to GRN163L, interruption of dosing until their platelet numbers returned to the normal range resulted in telomere re-elongating during the drug holiday period.
GRN163L (Imetelstat), is the first telomerase inhibitor in clinical trials, and 17 studies have been completed, including 10 phase 1 and 6 phase 2 trials. These are summarized in Table 3. The preliminary data in 50- to 74-year-old patients with advanced solid tumors (6 colorectal, 3 lung, 2 mesothelioma, 2 pancreas, 7 others) showed aPTT prolongation (active prolonged thromboplastin), gastrointestinal side effects, fatigue, anemia, GGT elevation, and peripheral neuropathy. Although thrombocytopenia is a dose-limiting toxicity at 4.8 mg/kg, one death from unknown reasons was observed at 3.2 mg/kg dose.88 Locally recurrent or metastatic breast cancer (MBC) patients were treated with GRN163L in combination with paclitaxel and bevacizumab (3 of 14 patients with de novo MBC and 6 of 14 patients with prior (neo) adjuvant taxane). Phase 1 clinical trials showed that the most common toxicities were cytopenia. The majority of patients had dose reduction or delays in treatments with GRN163L and/or paclitaxel owing to neutropenia and/or thrombocytopenia or other adverse effects.73 Main outcome from pediatric patients with solid tumors in phase 1 clinical trials was that the reduction of telomerase activity in PBMCs (peripheral blood mononuclear cells) and telomerase-positive tumors with the recommended phase 2 dose, which is 285 mg/m2 (MTD, maximum tolerated dose). In this study intravenous injection was conducted for 2 h on days 1 and 8 of a 21-day cycle. As a result, dose-limiting toxicity levels (DLT; 360 mg/m2) caused myelosuppression leading to a delay in the therapy of more than 14 days. Some patients had anemia, lymphopenia, neutropenia, thrombocytopenia, and catheter related infections depending on their grade level and cycle number of treatment.89 The advanced breast cancer phase 2 trial (NCT01256762) was stopped owing to poorer overall survival in patients treated with GRN163L.49 As GRN163L is administered on days 1 and 8 of a 21-day cycle, it is possible that telomerase cannot be inhibited sufficiently during this schedule, so that a new telomere equilibrium length for cancer cells emerges.
Table 3 GRN163L in clinical trials
| ClinicalTrials.gov Identifier/phase | Condition | Drug intervention | Status |
| NCT01568632/phase 1 | Refractory or recurrent solid tumors/lymphoma | GRN163L | Withdrawn |
| NCT01273090/phase 1 | Refractory or recurrent solid tumors/lymphoma | GRN163L | Completed |
| NCT01243073/phase 2 | Essential thrombocythemia/polycythemia vera (ET/PV) | GRN163L, standard of care | Suspended |
| NCT00594126/phase 1 | Refractory or relapsed multiple myeloma | GRN163L | Completed |
| NCT01916187/phase 1 | Neuroblastoma | GRN163L, 13-cis retinoic acid | Withdrawn |
| NCT00732056/phase 1 | Recurrent or metastatic breast cancer | GRN163L, paclitaxel, bevacizumab | Completed |
| NCT00310895/phase 1 | Refractory or relapsed solid tumor malignancies | GRN163L | Completed |
| NCT00718601/phase 1 | Multiple myeloma | GRN163L, bortezomib, dexamethasone | Completed |
| NCT01242930/phase 2 | Multiple myeloma | GRN163L, standard of care | Active, not recruiting |
| NCT00510445/phase 1 | Advanced or metastatic nonsmall-cell lung cancer | GRN163L, paclitaxel, carboplatin | Completed |
| NCT01265927/phase 1 | Her2+ breast cancer | GRN163L, trastuzumab | Active, not recruiting |
| NCT00124189/phase 1 | Chronic lymphoproliferative disease (CLD) | GRN163L | Completed |
| NCT02011126/phase 2 | Relapsed or refractory solid tumors | GRN163L | Withdrawn |
| NCT01137968/phase 2 | Nonsmall-cell lung cancer | GRN163L, bevacizumab | Completed |
| NCT01731951/Not provided | Primary or secondary myelofibrosis | GRN163L | Active, not recruiting |
| NCT01256762/phase 2 | Recurrent or metastatic breast cancer | GRN163L, paclitaxel, w/wo bevacizumab | Completed |
| NCT01836549/phase 2 | Recurrent or refractory brain tumors | GRN163L | Recruiting |
Challenges for developing telomerase inhibitors
The other potential challenge for telomerase inhibitors, such as GRN163L, is that the response time that is needed to induce senescence or crisis is long as telomere attrition occurs following each cell replication cycle. Therefore, this delayed response limits its utility as a first-line treatment for advanced cancer. It has been suggested that telomerase inhibitors following conventional therapy might be a good option to block the growth of residual cancer cells.65 A phase 2 study used GRN163L in a maintenance therapy setting after initial standard of care doublet chemotherapy for advanced nonsmall-cell lung cancer (NSCLC) has been completed (clinicaltrials.gov: NCT01137968). This study was designed to evaluate whether or not GRN163L, given as maintenance therapy, prolonged progression-free survival (PFS) in NSCLC. Eligible stage IV or recurrent locally advanced NSCLC patients were randomized to GRN163L (9.4 mg/kg over a 2-h IV infusion on day 1 and day 8 of each 21-day cycle) with or without bevacizumab on day 1 of each 21-day cycle, vs. standard of care alone (patients who had received bevacizumab before randomization were continued with bevacizumab maintenance or had no follow-up maintenance treatment). This study, while not achieving statistical significance showed a trend in PFS in the GRN163L arm, most apparent in the cohort of patients with the shortest telomeres at the initiation of the trial.90 Experience with these types of trials is important to more fully understand how to provide antitelomerase therapies going forward.
Overexpression of mutant hTERC and wild-type hTERC-targeted siRNA by lentiviral vectors are different types of gene therapy strategies for telomerase-targeted therapies
The other strategy to disrupt telomere maintenance in cancer cells is to express mutant hTR (hTERC) in the template region (mutant-template human telomerase RNA; MT-hTERC). Wild-type telomere repeats contain DNA binding sites for direct telomeric binding proteins such as POT1, TRF1, and TRF2 in humans.91, 92 It is predicted that mutated DNA that is directly synthesized by MT-hTERC disrupts the binding of telomeric proteins,93–95 which might lead to telomere uncapping. Studies showed that the uncapping of human telomeres by mutating TRF2 can induce growth arrest (p53 dependent) and apoptosis in cells.96, 97 Introduction of MT-hTERC via lentiviral vectors into telomerase-positive human cancer cells causes an alteration in the cancer cellular phenotype and reduced tumor growth in xenografts.94, 95 In another approach using siRNA (a hairpin short-interfering RNA) expressed from a lentiviral vector-targeted wild-type human telomerase RNA (WT-hTERC) also caused cell growth inhibition and apoptosis (these effects are telomere length independent). Coexpression of MT-hTERC and anti-WT-hTERC-siRNA showed additive or perhaps synergistic effects on cancer cells. Therefore, mutant hTERC and siRNA present promising anticancer therapy strategies owing to the lack of dependency on p53, initial telomere length, or progressive telomere shortening.98 However, these are still gene therapy approaches and require high efficiency in targeting to the cancer cells to be effective in the clinical setting.
6-Thio-dG is a small molecule that is a telomerase-based telomere uncapping target that may overcome some of the problems with gene therapy
Ideal cancer targets are those that are expressed in cancer cells, but not in normal cells. Compared to other targets, telomerase is one of the most universal targets expressed in almost all tumor cells, but not in normal cells except some proliferative progenitor stem cells. Therefore, telomerase is an important target to focus on for new therapeutic strategies.
So far, there have been two different therapeutic strategies in telomere biology: telomere or telomerase-targeted strategies. The strategy that targets telomerase inhibition is correlated with telomere length, whereas telomere-targeted approach is independent from initial telomere length (Figure 3). This makes an important difference between these two approaches. With only telomerase inhibition, a long lag period is expected (the time between enzyme inhibition and biological effect). Thus treatments will generally be long and depend on significant reduction of telomere length before reduction of tumor growth, senescence, or apoptosis is observed.29, 30
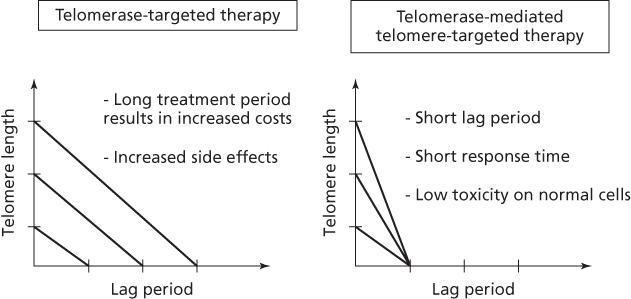
Figure 3 Telomerase-targeted therapeutic responses (such as Imetelstat/GRN163L) are dependent on the initial tumor cell telomere length. Cancer cells have variable telomere lengths. Therefore, cells with long telomeres are predicted to take a longer treatment period to be sensitive to telomerase inhibitors compared to cells with shorter telomeres (left side). As a result, any long-term treatment for telomerase may lead to unwanted side effects, high costs, and potentially re-establishing feedback loops establishing a stable new telomere length. In contrast to telomerase-targeted therapies, telomerase-mediated telomere targeting therapies (such as 6-thio-dG or MT-hTERC gene therapy) do not need long treatment periods as they are independent of the initial telomere length, therefore short response time to tumor burden reduction is a key advantage of these approaches (right side). The other most important advantage is that they may be less toxic to normal cells as most do not express telomerase. However, the MT-hTERC approach has the same obvious limitations of all gene therapy approaches of needing to reach almost all cells and delivery concerns. However, new approaches such as 6-thio-dG nucleoside may be orally available with minimal side effect and have the obvious advantage that all small molecules have.
Some approaches that target telomeres (not directly telomerase) lead to telomere uncapping (e.g., G-quadruplex), which is independent of p53 status and initial telomere length. However, normal tissue toxicity remains a major concern owing to quadruplex formation in other regions of the genome and in normal cells.99 Thus, approaches that do not target through telomerase may not have a therapeutic window of efficacy. In addition, using direct telomerase inhibitors or telomere-targeted therapies may need to be combined with other cytotoxic drugs to reduce the potential resistance capacity of heterogeneous tumor cells.99 For example, the cells from mTR knock-out mice showed telomere dysfunction by sensitizing cells to some anticancer drugs (e.g., doxorubicin).100 This result shows that telomerase-targeted therapy in combination with other chemotherapeutic agents may sensitize cells to other chemotherapeutics by resulting in reduced resistant capacity.
In recent work, we focused on a novel approach to identify a small molecule that would uncap telomeres but only in telomerase-positive cells. This theoretical approach using a small molecule obviates some of the problem with gene therapy and also could reduce the lag phase from initiation of treatment to tumor burden reduction. To test this concept, we decided to focus on nucleoside analogs that are good substrates for telomerase (not inhibitors of telomerase) and also more specific for telomerase than for other polymerases. 6-Thio-2′deoxyguanosine (6-thio-dG) is a new telomere-targeted nucleoside analog that is recognized by telomerase and is used by telomerase as a substrate.101 In our recent studies, we showed that 6-thio-dG causes telomere dysfunction by causing altered telomere structures called telomere dysfunction-induced foci (TIFs) in telomerase-expressing cancer cells of varying telomere lengths and comparing these to normal and ALT cells. Interestingly, beside short-term cell killing effects, 6-thio-dG caused rapid and progressive telomere shortening in surviving cancer cells but not in normal cells. These studies demonstrated that 6-thio-dG targets telomeres by being preferentially used by telomerase as a substrate. In human xenograft models in immunodeficient mice, 6-thio-dG reduced lung cancer growth (no observed side effects, e.g., no death, no weight loss) and caused telomere dysfunction. There were no long-term side effects on normal tissues and hematological, liver, and kidney functions were in the acceptable range. Thus, 6-thio-dG is a promising “telomeric poison” targeting telomeres by using telomerase as a catalyst to change telomeres so they are recognized as DNA damage. Additional, perhaps more potent nucleoside analogs will be the new direction for targeting the cell immortality cancer hallmark.
Summary
Telomeres are protective structures that are found at the ends of linear chromosomes. Telomeres shorten with each cell division owing to the end replication problem. However, telomerase maintenance mechanisms can counteract this progressive shortening process. While most normal cells do not express telomerase activity, it is detected in almost all primary human tumors. Therefore, telomerase is a unique and almost universal target for cancer.
Although there have been promising approaches targeting telomerase for cancer therapies, there are no drugs that have been approved. G-quadruplex stabilizers that target the G-rich DNA sequences in human telomeres cause telomerase inhibition and telomeric end disruption. However, the specificity of these compounds is very limited and may affect quadruplex structures in normal cells as well as cancer cells.
Immunotherapy approaches in clinical trials utilize hTERT GV1001 (peptide vaccine) and GRNVAC1 (dendritic cell priming ex vivo). The utility of these vaccines in combination with conventional therapy may be limited owing to immunosuppressive effect of chemotherapeutic drugs.
GRN163L (Imetelstat) is a telomerase-targeted oligonucleotide approach. GRN163L shows highly potent telomerase inhibition, resulting in progressive telomere shortening in cancer cells. Owing to adverse hematological events, clinical trials in solid tumors have not progressed. One of the potential challenges for GRN163L is the response (long lag) time that is needed to induce senescence/apoptosis.
Gene therapy is a well-established technique that targets cancer by delivering therapeutic DNA. Mutant hTERT (dominant-negative approach) or siRNA directed at hTERT are some of the different gene therapy approaches in preclinical studies. In addition, introduction of a mutant hTERC via lentiviral transfer is a telomerase-dependent telomere “upcapping” approach. Gene therapy is a promising anticancer therapy approach but requires high efficiency to target the vast majority of cancer cells.
Typical telomerase inhibitors (GRN163L) require a long lag period between enzyme inhibition and biological effect. In contrast, telomerase-mediated telomere-targeted therapy is predicted to have a much shorter lag period. One example is 6-thio-dG, a nucleoside analog. 6-thio-dG is a new approach to target telomeres, is preferentially recognized by telomerase and incorporated into telomeres resulting in TIFs. This small-molecule approach results in short-term cell killing effects on telomerase-expressing cells (short lag time) with minimal effects on telomerase silent normal cells (e.g., quiescent stem cells).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


