INTRODUCTION
SUMMARY
The acute inflammatory response is characterized by a rapid but relatively short-lived localized increase in blood flow, an increase in microvascular permeability and the sequential recruitment of different types of leukocytes. Acute inflammation may be followed by “chronic” inflammation and a superimposed series of reparative processes (e.g., angiogenesis, production of extracellular matrix, parenchymal regeneration and scar formation). The early hemodynamic changes at a site of inflammation establish low shear conditions that enable marginated leukocytes to engage in low-affinity selectin-mediated rolling interactions with activated endothelial cells. In response to locally produced soluble and cell surface mediators, endothelial cells and rolling leukocytes sequentially express several sets of complementary adhesion molecules that include selectins, integrins, and members of the immunoglobulin superfamily. Leukocyte and endothelial cell adhesion molecules mediate the high-affinity adhesive interactions necessary for leukocyte emigration from the vascular space along chemotactic gradients. Analogous, temporally regulated, soluble mediators and cellular adhesion molecules also orchestrate succeeding monocyte- and lymphocyte-rich chronic inflammatory responses. This paradigm is modulated by a vast network of surface-active and soluble inflammatory mediators. Recruited leukocytes and cells indigenous to the anatomic site of inflammation both play critical roles in host defense, resolution of inflammation and tissue repair.
Acronyms and Abbreviations:
ADAM, a disintegrin and metalloproteinase; BPI, bacterial permeability-increasing protein; CAP37, cationic antimicrobial protein; DARC, Duffy antigen receptor for chemokines; CD, cluster of differentiation; eNOS, endothelial nitric oxide synthase; HEV, high-endothelial venule; HPETE, hydroperoxyeicosatetraenoic acid; ICAM, intercellular adhesion molecule; IFN, interferon; Ig, immunoglobulin; IL, interleukin; iNOS, inducible nitric oxide synthase; LT, leukotriene; LTB4/C4/D4/E4, leukotriene B4/C4/D4/E4; MadCAM, mucosal addressin cell adhesion molecule; MASP, mannan-binding lectin-associated serine protease; MBL, mannan-binding lectin; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); NO, nitric oxide; PAF, platelet-activating factor; PARs, proteinase-activated receptors; PNAd, peripheral node addressin; PSGL-1, P-selectin glycoprotein ligand-1; RGD, arginine-glycine-aspartic acid peptide sequence; TACE, tumor necrosis factor-α converting enzyme; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule; VLA, very-late antigen.
HISTORY
The sentinel clinical features of acute inflammation—rubor, calor, tumor, and dolor—have been recognized for at least 5000 years.1 Dr. John Hunter, the renowned late 18th-century Scottish surgeon, observed that the inflammatory response is not a disease per se but rather a nonspecific and salutary response to a variety of insults. Through his microscopic examinations of transparent vital membrane preparations, German pathologist Julius Cohnheim concluded that the inflammatory response is fundamentally a vascular phenomenon. Phagocytosis was described late in the 19th century by Elie Metchnikoff and his colleagues at the Pasteur Institute. Morphologic studies, using both live animals and fixed histologic preparations, transformed our understanding of inflammation and led to the currently held concepts of inflammation-associated hemodynamic alterations, acute inflammation and chronic inflammation.1,2 During the past 5 decades, the modern techniques of biochemistry, tissue culture, monoclonal antibody production, recombinant DNA technology, and the genetic manipulation of isolated cells and whole animals have enabled a more detailed understanding of the cellular and molecular mechanisms which underpin the inflammatory response, the active resolution of inflammation, and the transition to tissue repair and restitution. These studies, in concert with “experiments of nature,” such as chronic granulomatous disease (Chap. 66) and the leukocyte adhesion deficiency disorders (Chap. 66), have permitted the formulation of complex, yet elegant, models of acute and chronic inflammation and led to the promise of incisive therapeutic approaches. A large array of human diseases is marked by either defects in the development of the inflammatory response or the deleterious effects of the inflammatory response itself.
GENERAL CHARACTERISTICS OF INFLAMMATION
It is useful to consider inflammation as an acute or chronic (persistent) process. Acute inflammation lasts from minutes to several days and is characterized by pronounced local hemodynamic and microvascular changes and leukocyte accumulation.2,3 The acute inflammatory response is consistently marked by microvascular leakage and the accumulation of neutrophils. The four cardinal signs of acute inflammation, alluded to above, can be accounted for within the physiologic parameters of inflammation. The systemic effects of inflammation, particularly acute inflammation, account for the familiar clinical findings of fever, acute-phase response and altered sensorium. In turn, sepsis is a systemic inflammatory response syndrome that occurs in response to infection; severe sepsis is sepsis complicated by acute organ dysfunction; and septic shock is sepsis complicated by either fluid resuscitation-resistant hypotension or by hyperlactatemia.4
The chronic inflammatory response, which lasts much longer and is more varied in its effects, is marked by growth of new capillaries and proliferation of resident fibroblasts.2,3 Cellular infiltrates typically include monocytes and lymphocytes, but there are many variations in the cellular composition, anatomic distribution and tempo of development of chronic inflammatory lesions. Chronic inflammatory processes can be classified according to these variations. For example, granulomatous inflammation is a chronic process marked by nodular aggregates of mononuclear phagocytes that have become “transformed” into epithelioid histiocytes, so-called because of their similar appearance to epithelial cells.2 Granulomas may be distributed along blood vessels (e.g., angiocentric), along upper airways (e.g., bronchocentric), or randomly throughout the interstitium or parenchyma of an organ. Granulomas may vary morphologically. Tuberculous granulomas often contain areas of caseous necrosis while sarcoidosis-associated granulomas are often cellular and exhibit fibrosis but usually without areas of necrosis.2 Other chronic inflammatory processes are marked by a preponderance of eosinophils or plasma cells. In contrast to the more stereotyped appearance of an acute inflammatory lesion, the particular appearance of a chronic inflammatory lesion can sometimes provide insight into its cause (e.g., caseating granulomas in tuberculosis, eosinophil-rich infiltrates in a parasitic infection and plasma cell-rich infiltrates in viral hepatitis).
Superimposed upon acute and chronic inflammatory responses is repair.2,5 Resolution or termination of the inflammatory response is an important step in the pathway to repair; it occurs through a complex set of regulated processes.5 Repair, which may entail the regeneration of parenchymal cells damaged as the result of an insult per se or as “bystanders” to the inflammatory response, is characterized by the growth of new capillaries (angiogenesis) and the activation of fibroblasts which produce extracellular matrix molecules (e.g., scar tissue). In some circumstances an inflammatory response is self-limited (e.g., sunburn), whereas in other situations the response may persist for many years (e.g., tuberculous granulomas). The elimination or persistence of an insult has a major influence on outcome–whether ongoing chronic inflammation, complete regeneration or scar formation. There is great complexity in terms of the networks of proinflammatory and antiinflammatory soluble mediators (e.g., cytokines) and the phenotypes, as well as the functional and regulatory roles of both indigenous cells and recruited inflammatory cells in inflammation, resolution and repair.2,3,5,6,7
This chapter first addresses acute inflammation, which encompasses localized changes in blood flow, alterations in microvascular permeability and neutrophil exudation.2,3 In addition to hemodynamic changes, inflammation encompasses endothelial cell activation, low-affinity leukocyte–endothelial adhesion, high-affinity or stationary leukocyte–endothelial adhesive interactions, leukocyte emigration, leukocyte activation, and the subsequent dampening and resolution of the inflammatory response. The highly regulated migration of leukocytes from the vasculature into sites of inflammation and of lymphocytes through secondary lymphoid tissues and, in turn, into sites of microbial invasion, are pivotal to host defense in the contexts of inflammation and immunity.2,3,6 This extraordinary complexity of regulatory processes that control inflammation is exemplified by, but not limited to, proinflammatory cytokines (e.g., tumor necrosis factor [TNF]-α, interleukin [IL]-1β, IL-6) that drive inflammation, countered by rises in antiinflammatory cytokines (e.g., IL-4, IL-10, IL-11, IL-13, transforming growth factor [TGF]-β, IL-1ra and soluble cytokine receptors) that dampen inflammatory responses (see “Cytokines and Chemolines”).2,7 Both the termination of an inflammatory process and the transition from an active inflammatory milieu to a wound-healing, tissue-remodeling environment are actively regulated processes. The concept of “active” termination of the inflammatory response, including the roles of chemokine depletion, neutrophil apoptosis, resolvins and protectins, and the shift from interferon (IFN)-γ–driven “classical M1” macrophages to IL-4/IL-13–driven “alternative M2” macrophages, is introduced at the end of the first section of this chapter (M1 and M2 Macrophages). The section “Regulators of the Inflammatory Response” of this chapter introduces (and where appropriate, reiterates) the vast array of soluble and surface-active mediators that regulate both acute and chronic inflammatory responses, as well as some aspects of resolution. These mediators include substances that range from short-lived reactive oxygen and nitrogen intermediates to entire regulatory systems (e.g., complement and coagulation). Many mediators of inflammation have become targets for therapeutic interruption strategies. See section “Chronic Inflammation and Repair.” This chapter provides a framework for understanding the basic processes of inflammation while promoting an appreciation for the highly complex and integrated nature of the regulated inflammatory response.
ACUTE INFLAMMATION
The hemodynamic changes that occur early in acute inflammation include arteriolar vasodilatation and localized increases in microvascular permeability (Fig. 19–1). In many circumstances, arteriolar vasodilation follows a rapid and transient period of vasoconstriction.2,3 Arteriolar vasodilation results in increased blood flow, thus explaining the familiar redness and warmth that characterize a site of acute inflammation. The increase in blood flow, coupled with increases in microvascular permeability, results in hemoconcentration and increased local viscosity. These hemodynamic changes are critical to subsequent leukocyte emigration because selectin-mediated low-affinity rolling leukocyte–endothelial adhesive interactions occur only under such conditions of low shear force. Experimental studies using in vitro flow chambers and transparent vital membrane preparations in live animals indicate that selectin-mediated leukocyte–endothelial rolling adhesive interactions cannot occur in the face of the shear forces that exist under conditions of normal blood flow velocity. Increased microvascular permeability leads initially to protein-poor transudation followed by protein-rich plasma exudation, another characteristic of acute inflammation.2 Microvascular leakage occurs through a variety of temporally regulated mechanisms, including rapid, reversible, and short-lived venular endothelial cell contraction attended by widening of intercellular junctions; so-called endothelial cell retraction, which is less-well understood but involves long-lived cytokine-mediated cytoskeletal changes; direct endothelial injury and disruption by physical trauma; leukocyte-mediated endothelial cell injury; and leakage via new capillaries that do not yet possess completely “closed” intercellular junctions.2 Increases in rate of transcytosis by which plasma constituents cross endothelial cells in vesicles or vacuoles (vesiculovacuolar organelles) occur in neoplastic blood vessels and may play a role in inflammation.2,8 Alterations in local blood flow occur at the level of arterioles, the key to vascular resistance and regulated largely by the autonomic nervous system, nitric oxide (NO) (formerly called endothelium-derived relaxing factor), vasoactive peptides, and eicosanoids. A variety of soluble mediators can induce increases in microvascular permeability through several of the above-mentioned mechanisms.
Figure 19–1.
Early hemodynamic events in acute inflammation. Vascular dilatation, increased microvascular permeability, fluid transudation, and leukocyte recruitment and emigration occur after a transient period of arteriolar vasoconstriction. (Modified and redrawn with permission from Cotran RS, Kumar V, Collins T, Robbins SL (eds): Robbins Pathologic Basis of Disease, 6th ed. Philadelphia, PA: Saunders/Elsevier; 1999.)
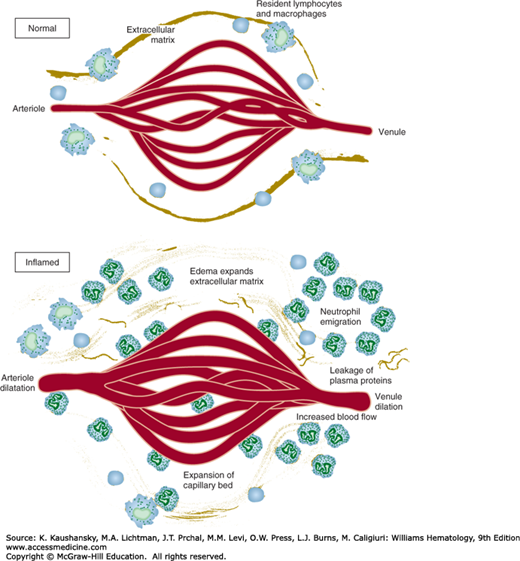
The recruitment of leukocytes into a site of inflammation is a fundamental characteristic of the inflammatory response.2,3,9 The orchestrated recruitment of particular types of leukocytes into specific tissues, whether sites of acute inflammation, in the course of physiologic lymphocyte recirculation through lymph nodes or in the cellular immune response to microbial invasion, is referred to as homing.3 The general mechanisms of leukocyte homing are similar, but the leukocytes and particular mediator molecules vary. For example, neutrophils bind and traverse postcapillary venules in acute inflammation, naïve T lymphocytes bind and traverse lymph node high-endothelial venules (HEVs) in lymphocyte recirculation, and effector and memory T lymphocytes bind and traverse postcapillary endothelial cells in sites of chronic infection.3 The importance of white blood cells in host defense is highlighted in patients with either numerical leukocyte deficiencies or functional defects. Leukocytes are critical because of their central role in the phagocytosis and killing or containment of microbes and in the digestion of necrotic tissue debris. Leukocyte-derived products, such as proteolytic enzymes and reactive oxygen intermediates, contribute to tissue injury.
Vascular stasis that results from the hemodynamic changes of early acute inflammation leads to displacement of leukocytes from the central axial column of circulating blood cells to positions along the endothelial surface. This process, margination, is enhanced under conditions of slow blood flow.2,3 Leukocytes adhere transiently and weakly to the endothelial surface. Vital membrane preparations and flow chamber studies using endothelial cell monolayers and suspensions of purified leukocytes have revealed that cells “tumble and roll” along the endothelial surface.10 Transient, weak, rolling neutrophil–endothelial adhesive interactions occur within minutes of initiation of an acute inflammatory response and can, depending upon the time point within the evolution of an inflammatory response, involve neutrophils, lymphocytes, monocytes, basophils, or eosinophils. Leukocyte–endothelial cell-rolling adhesive interaction is a specific and necessary step that precedes high-affinity, or so-called stationary adhesion and emigration.9,10,11 Early rolling adhesive interactions are mediated largely by selectins and their carbohydrate-rich counterreceptors.2,3,9,10 In turn, the cell-surface expression of selectins (and other intercellular adhesion molecules) is regulated by locally produced proinflammatory mediators.9,10,11
Selectins contain an extracellular N-terminal carbohydrate-binding region that is homologous to mammalian lectins, an epidermal growth factor-like domain, a series of complement regulatory domains, and a lipophilic transmembrane domain (Table 19–1).2,3,9,10 P-selectin is expressed by endothelial cells and platelets, E-selectin by endothelial cells, and L-selectin by most white blood cells. P-selectin is constitutively synthesized and stored in endothelial intracytoplasmic granules (Weibel-Palade bodies).9 When endothelial cells are exposed to histamine, thrombin, platelet-activating factor (PAF) or tissue factor, preformed P-selectin is rapidly (within minutes) translocated to the endothelial surface where it engages marginated leukocytes via carbohydrate moieties that contain sialic acid residues (e.g., P-selectin glycoprotein ligand-1 [PSGL-1]).2,3,10,12 This transient, low-affinity, binding interaction accounts in part for the early rolling interactions (Fig. 19–2). The development of single knockout mice (i.e., lacking individual selectins [P−/−; E−/− or L−/−], double knockout mice (e.g., E−/− and P−/−), and even triple knockout mice, has confirmed that leukocyte rolling can be almost completely accounted for by selectins.13,14 Exposure of endothelial cells to TNF-α or IL-1β results in new protein synthesis–dependent expression of E-selectin, a response that occurs within 1 to 2 hours and peaks at 4 to 6 hours.2,3,9 As in the case of P-selectin–mediated leukocyte adhesion, E-selectin–mediated adhesion occurs via a series of sialylated and fucosylated carbohydrate moieties related to the sialyl Lewis X and sialyl Lewis A blood group antigens, but found on leukocytes (Table 19–1).9,10 Selectin counterreceptors consist of several different mucin-like glycoproteins coated with various sialyl moieties. L-selectin is constitutively expressed by leukocytes, participates in neutrophil and monocyte binding to activated endothelium in inflammatory sites, lymphocyte–endothelial cell homing (e.g., lymphocyte homing to lymph nodes via HEVs), leukocyte–leukocyte adhesive interactions via sulfur-containing mucin-like glycoproteins, and is cleaved from cells by means of “sheddase” enzymes such as a disintegrin and metalloproteinase (ADAM)-17 (TNF-α converting enzyme [TACE]) when the leukocyte is activated (see Table 19–1).3,15 The relevant mucin-like glycoprotein counter-receptors on HEVs are collectively referred to as peripheral node addressins (PNAds) and include mucosal addressin cell adhesion molecule (MadCAM)-1, GlyCAM-1, and CD34.3,15 L-selectin shedding facilitates leukocyte emigration by allowing the weakly adherent white blood cell to detach from the endothelium. Low-affinity rolling adhesive interactions set the stage for β-integrin– and immunoglobulin superfamily mediated high-affinity adhesive interactions and leukocyte transmigration.2,3,9
| Family | Structure | Members | Tissue Distribution | Counterreceptor* |
|---|---|---|---|---|
| Selectin | N-terminal lectin domain, epidermal growth factor domain, multiple complement regulatory repeats, transmembrane, and short cytoplasmic tail | P-selectin | Endothelium, platelets | PSGL-1, SLex glycoprotein |
| E-selectin | Endothelium | PSGL-1, SLex glycoprotein | ||
| L-selectin | Leukocytes | PNAds: GlyCAM-1, MadCAM-1, CD34 | ||
| Immunoglobulin superfamily | Multiple immunoglobulin domains, transmembrane region and cytoplasmic tail | ICAM-1 | Endothelium, other cells | CD11a/CD18 |
| ICAM-2 | CD11b/CD18 | |||
| ICAM-3 | ||||
| VCAM-1 | Endothelium | VLA-4 | ||
| CD31 (PECAM) | Endothelium | CD31 | ||
| Integrin (β2; leukocyte) | Heterodimers: distinct α subunits with common β subunits | CD11a/CD18 (LFA-1) | Neutrophils, monocytes, macrophages, and lymphocytes | ICAM-1 |
| ICAM-2 | ||||
| ICAM-3 | ||||
| CD11b/CD18 (Mac-1) | Neutrophils, monocytes, and macrophages | ICAM-1, iC3b, LPS, and fibronectin | ||
| VLA-4 | Monocytes and lymphocytes | VCAM-1 and fibronectin |
Figure 19–2.
Leukocyte–endothelial adhesive interactions. Early in the acute inflammatory response, marginated leukocytes engage in transient, low-affinity, selectin-mediated rolling adhesive interactions with endothelial cells. As the response evolves, activated leukocytes and endothelial cells engage in high-affinity, β2-integrin– and immunoglobulin superfamily mediated adhesive interactions. A variety of chemotactic factors trigger the motive force for leukocyte emigration.
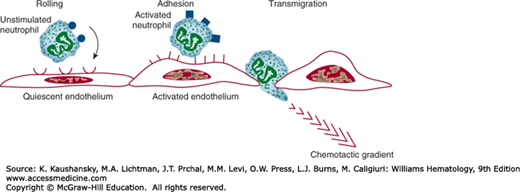
Weak selectin-mediated rolling and high-affinity stationary adhesive interactions are not temporally or mechanistically completely discrete. For example, TNF-α and IL-1β both induce E-selectin, which is not expressed by quiescent cells, and both increase endothelial expression of intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, which are constitutively expressed in low cell-surface densities.2,3 ICAM-1 is involved in the recruitment of all types of leukocytes and VCAM-1 is involved in the recruitment of chronic inflammatory leukocytes (lymphocytes, monocytes, eosinophils, and basophils).2,3,9 ICAM-1 binds to β2 (leukocyte) integrins, which are heterodimeric structures that contain one species of α chain (e.g., CD11a, CD11b, CD11c, CD11d) and a common β chain (CD18).16 VCAM-1 binds to β1 integrins (e.g., very-late antigen [VLA]-4/α4β1) (see Table 19–1).2,3 Activated endothelial cells secrete PAF and CXCL8 (IL-8), which activate overlying selectin-bound leukocytes.2,3,9,16 Individual leukocyte CD11a/CD18 (LFA-1) heterodimers undergo a transient conformational change and groups of CD11a/CD18 molecules form multimolecular clusters.2,3,9 Both the conformational change in CD11a/CD18 and the clustering contribute to increases in binding affinity to endothelial ICAM-1.3,16 CD11b/CD18 (Mac-1) binds ICAM-1, ICAM-2, and iC3b (see section “Complement”), the latter of which opsonizes complement-coated particulates. CD11c/CD18 also binds to iC3b and initiates phagocytosis, but plays a lesser role in neutrophil adhesion than do CD11a/CD18 and CD11b/CD18. Intercellular adhesion molecules are found on a variety of cell types other than endothelial cells. The roles of CD11c/CD18, CD11d/CD18, and ICAM-3 in leukocyte–endothelial adhesion are less-well established. β1-Integrins, notably VLA-4, are found primarily on chronic inflammatory leukocytes (e.g., lymphocytes, monocytes, basophils, and eosinophils) and mediate leukocyte binding via VCAM-1.2,3,9 β1-Integrin–mediated adhesive interactions occur via arginine-glycine-aspartic acid peptide sequences (RGDs) displayed by VCAM-1, as well as on exposed surfaces of matrix molecules (e.g., fibronectin). β2-Integrin–ICAM-1 and β1-integrin–VCAM-1–mediated adhesive interactions occur later (hours to days) in the inflammatory response than do selectin-mediated interactions.
β1– and β2-integrins are clearly important in leukocyte recruitment into sites of acute and chronic inflammation. Eighteen different integrin α subunits, eight integrin β subunits, and 24 in vivo heterodimer combinations have been identified in mammals.17 In addition to leukocyte–endothelial cell adhesion interactions, integrins function in a variety of other cell–cell and cell–extracellular matrix interactions. A large number of integrin-targeted small molecule, peptide, and designer antibodies have been developed for therapeutic applications.17 Inflammatory–immunologic diseases treated with integrin antagonists include, among others, multiple sclerosis, Crohn disease, and age-related macular degeneration.
High-affinity stationary adhesive interactions precede leukocyte transmigration across the endothelium into the subjacent interstitium. The functional importance of complementary leukocyte–endothelial adhesive interactions has been clarified by in vitro binding studies and in vivo studies that have employed neutralizing antibodies directed against adhesion molecules, pharmacologic antagonists of adhesion molecules, and knockout mice.2,3 The functional importance of leukocyte integrins (CD11a/CD18, CD11b/CD18, CD11c/CD18) has also been highlighted by clinical and experimental observations in patients with rare genetic leukocyte adhesion deficiencies (Chap. 66).
β2-Integrin ICAM-1 and ICAM-2, as well a β1-integrin (VLA-4) VCAM-1, induce adhesive interactions that lead to cytoskeletal reorganization in leukocytes that flatten and spread out on the endothelial surface, extend pseudopodia between endothelial cells, and migrate along extravascular chemotactic gradients. Most leukocytes exit the vascular space between adjacent endothelial cells (paracellular transmigration). Paracellular transmigration depends not only on integrin-ligand interactions but also on CD31 (PECAM-1 [platelet-endothelial cell adhesion molecule-1]) expressed on both leukocytes and endothelial cells, and transient reversible disassembly of tight interendothelial vascular endothelial (VE)–cadherin junctional complexes.18 There is evidence that leukocytes can also exit the vascular space via a less-well-characterized transcellular pathway.
Leukocytes bound tightly to endothelium emigrate from the vascular space into the interstitium by extending pseudopods between intercellular junctions (see Fig. 19–2).18 Secreted neutral proteases such as elastase, cathepsin G, and proteinase 3, play a role in the passage or “invasion” of leukocytes through the subendothelial extracellular matrix. Collagenases are particularly important in leukocyte transmigration through basement membranes. A variety of matrix metalloproteinases, produced by several cell types, participate in leukocyte migration, and also play roles in resolution of inflammation and tissue remodeling. Leukocyte emigration and subsequent movement through the interstitium follow chemical concentration gradients; processes facilitated by binding interactions between leukocyte integrins and complementary sites on extracellular matrix molecules (e.g., fibronectin).9 A wide variety of soluble mediators can provide the motive trigger for this process.2,3,19 Chemotactic factors for neutrophils include peptides derived from bacteria (e.g., N-formyl peptides), complement-derived peptides (e.g., C5a), cell membrane-derived chemotactic lipids (e.g., PAF), and cytokines and chemokines produced by a variety of cell types (e.g., CXCL8 [IL-8] from endothelial cells).2,3,19 Chemotactic factors vary with respect to their specificity for different types of leukocytes. For example, C5a and N-formyl peptides both induce neutrophil and monocyte chemotaxis, and CXCL8 [IL-8] induces neutrophil chemotaxis, whereas CCL2 (monocyte chemoattractant protein [MCP]-1) induces chemotactic responses in monocytes and a specific subset of memory T lymphocytes.3 The Th17 subset of CD4 T-helper lymphocytes secretes IL-17 and IL-22 which participate in the recruitment of neutrophils.3 (There are several species of IL-17, including homodimers IL-17A–IL-17F as well as heterodimeric species.20) Each of these chemotactic factors activates “target” cells by engaging specific cell surface receptors, which, in turn, are linked to the contractile cell motility apparatus.3,19
In addition to chemotaxis, soluble and cell-surface mediators induce leukocyte activation manifested by a wide array of changes in cellular function (e.g., leukocyte integrin upregulation and increased binding affinity [e.g., CD11b/CD18], selectin shedding [e.g., L-selectin], lysosome degranulation, and initiation of the respiratory burst). There have been great advances in understanding of the biochemical pathways involved in chemotaxis, cell activation, and degranulation.21 Although there are many nuances in the signal transduction pathways involved in these processes, several themes have emerged. Cell surface receptors are activated by specific ligands (e.g., C5a, leukotriene B4 [LTB4], CXCL8 [IL-8] and receptor activation is transduced via specific G proteins and membrane-associated phospholipases, which leads to mobilization of intracellular calcium, influx of extracellular calcium and phosphorylation of series of cytosolic proteins.19 Rare genetic diseases linked to receptor and effector defects (e.g., IFN-γ receptor defects and nicotinamide adenine dinucleotide phosphate [reduced form] [NADPH] oxidase defects) have provided insight into leukocyte function and the importance of such specific activities in host defense (Chap. 66).
A principal result of neutrophil and monocyte recruitment is provision of large numbers of activated leukocytes that can release lytic substances and reactive oxygen and nitrogen intermediates needed to destroy foreign invaders, and a vehicle to contain foreign particulates through phagocytosis. Some recruited monocytes differentiate into macrophages and recruited effector and memory lymphocytes play pivotal roles in the adaptive immune response.3,6 The products and functions of activated inflammatory cells are at once salutary because they contain and destroy invaders and deleterious because they cause tissue damage. The roles of neutrophil apoptosis (programmed cell death) in the termination of acute inflammatory responses and IFN-γ–driven M1 macrophages and IL-4/IL-13–driven M2 macrophages in the transition of an inflammatory milieu to a wound-healing or tissue-remodeling milieu are discussed in “M1 and M2 Macrophages”.
Leukocyte activation, especially of neutrophils and mononuclear phagocytes, results in the secretion of microbicidal peptides (e.g., defensins, bactericidal permeability-increasing protein [BPI], cationic antimicrobial protein [e.g., CAP37]) and lytic enzymes (e.g., myeloperoxidase, elastase, cathepsin G).6,21,22 The release of such granular constituents is accompanied by the generation of reactive oxygen and nitrogen intermediates (e.g., O2−, H2O2, NO), the generation of arachidonate metabolites (e.g., leukotrienes and prostaglandins) and the production of other proinflammatory mediators (see section “Regulators of the Inflammatory Response”).21,22 In some circumstances these mediators are released into phagolysosomes where they contribute to the destruction of engulfed microbes, while in other circumstances they are secreted into the extracellular milieu where they amplify the inflammatory response and cause tissue damage. The various types of neutrophil granules (primary azurophilic, secondary specific, tertiary gelatinase-containing, and secretory vesicles) are released in a differentially coordinated fashion.21,22
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


